珠海防護用品消毒劑檢測?美國醫療器械行業為何發展的這么好 健明迪檢測
珠海防護用品消毒劑檢測

防護用品CE認證:口罩檢測規范:EN149,普通防護服檢測規范:EN13688,一次性化學防護服檢測規范:EN14325,護目鏡檢測規范:EN166。 檢測可以依據國度規范、ISO規范、美國ASTM規范、歐盟EN規范等相關規范,為您提供專業的檢測,圍繞物理功用、拉伸功用、化學功用、生物學評價等方面停止專業測試。
● 測試范圍 ●
口罩、呼吸防護用品、醫用防護服、醫用面部防護產品、消毒劑質量檢測/備案申報、醫用防護手套、防護產品原資料等。
● 檢測項目 ●
消毒劑檢測項目:外觀、有效成分含量測定、pH值測定、動搖性實驗、延續運用動搖性實驗、鉛、砷、汞的測定、金屬腐蝕性實驗、實驗室對微生物殺滅效果測定、模擬現場實驗或現場實驗、毒理學平安性檢測、總體功用實驗。
抑菌劑抗菌劑項目:有效成分含量測定、動搖性實驗、pH值測定、微生物目的、 細菌菌落總數、大腸菌群、真菌菌落總數、致病性化膿菌。
衛生用品檢測項目: 大腸桿菌殺滅(抑制)實驗 、金黃色葡萄球菌殺滅(抑制)實驗 、白色念珠菌殺滅(抑制)實驗 、其他微生物殺滅(抑制)實驗 、毒理學目的檢測。
口罩檢測項目:理化目的、微生物目的 、毒理學過敏性目的。
防護產品檢測項目:功用檢測,防護等級檢測,微生物檢測,氣密性檢測,防火等級檢測,平安性檢測,運用壽命評價檢測,老化檢測,防塵檢測,面料檢測,質量檢測,出廠檢測,色牢度檢測,熄滅功用檢測,抗穿刺功用檢測,撕裂功用檢測等。
珠海防護用品消毒劑檢測?美國醫療器械行業為何開展的這么好? 健明迪檢測
潔凈室對高效過濾器泄露測試的要求

在市場概述局部,我們描畫了污染物對人、產品和環境的影響。在關鍵的消費環境中,污染物會招致產品損壞、產量下降、產品召回以及代價高昂的業務中綴。污染控制有利于維護航空航天、微電子、制藥、醫療器械、醫療保健和食品以及核能等行業運用中的產品或進程完整性。無論是在生命迷信(病原微生物)、彌補效勞(石棉、霉菌)還是核工業(輻射)范圍,工人都需求維護免受污染。 污染物會以多種方式經過微粒滲入消費環境區域 · 乘氣流出境 · 工人的工棚 · 從環境、設備或資料中零落 · 或許在外部環境和關鍵區域外部之間移動的任何載體的腳上。 行業規范規則了空氣清潔度,即空氣中的顆粒數量,以及確定清潔度的測試方法,包括采樣位置的選擇。 將污染控制歸入其規范協議的*罕見環境之一是潔凈室和清潔區. 潔凈室 潔凈室是一個受控的環境,其建造和運轉方式限制了室內空氣傳達顆粒的引入、發生和滯留。 經過向房間/區域提供少量空氣來控制顆粒,這些空氣曾經過高效過濾器的過濾。這種空氣有兩個功用: (1)稀釋和去除房間內人員和機器散布的顆粒和細菌, 2)給房間加壓,確保沒有臟空氣流入潔凈室。 潔凈室由不發生顆粒且易于清潔的資料建造,嚴厲的規程要求任務人員運用防護服,以*限制地增加顆粒和微生物的分散。潔凈室還控制溫度、濕度、聲響、照明、靜電放電和振動,以進一步維護產品和在其中任務的人員。 清潔區 潔凈區是一個確定的空間,在該空間內,空氣傳達顆粒的濃度失掉控制和分類。潔凈區可以是潔凈室中確定的空間,也可以經過位于潔凈室外部或外部的分別裝置來完成。 分別或清潔空氣裝置 分別裝置是堅持一定體積內外分別的設備。分別是經過裝置的結構和氣流完成的。 大少數分別設備在潔凈室的關鍵區域提供高質量的過濾空氣。清潔空氣裝置的類型包括單向氣流柜和外殼、限制進入屏障系統(RABS)、隔離器、微型環境和生物平安柜。 單向氣流柜(非過濾分別裝置) 單向氣流柜(也稱為通風櫥或層流柜或任務臺)是*復雜的分別設備,主要用于人體接觸不會形成污染的狀況。 機柜將未過濾的廢氣吹向用戶,關于病原體的任務是不平安的。當工人坐在任務臺上處置資料時,空氣流向人和任何污染物(死皮細胞等)。)由人分發到空氣中,堅持在關鍵進程的下風處。 這些設備主要用于防止半導體晶片、生物樣品或任何顆粒敏感資料的污染,但不會增加工人接觸正在處置的資料時的接觸污染。單向氣流設備雖然復雜,但可將空氣中的顆粒和微生物污染增加至少10倍,*多100倍或更多。 生物平安柜(一種過濾分別裝置) 生物平安柜是一個封鎖、通風的實驗室任務空間,用于平安地處置被病原體污染的資料,需求規則的生物平安等級。 經過該室再循環的空氣經過供氣HEPA過濾器,在該室內構成一個無菌環境,去除有害物質,如病毒和細菌。 照片顯示一名迷信家在生物平安柜中任務,通風櫥外的空氣被吸入柜中,然后在再循環到實驗室大氣中之前過濾掉任何病原體。圖片來源:James Gathany內容提供商:CDC -該媒體來自疾病控制和預防中心 公共衛生圖像圖書館(菲爾),帶識別號#7988. 生物平安柜的等級 有三種生物平安柜。 一級為用戶和周圍環境提供維護,但不為被操作的樣品提供維護。空氣被拉過開放的任務區,并在排氣口裝置了HEPA過濾器。I級機柜用于對機柜內的環境能否比周圍環境更潔凈沒有嚴厲要求的中央。 第二類柜體(左下方)用于柜體內的條件比周圍環境更潔凈的重要場所,并為用戶、環境和樣品提供維護。HEPA過濾任務區上方的垂直向下氣流,前啟齒處的向內氣流,HEPA過濾排氣。二級平衡計分卡分為四種類型:A1、A2、B1和B2。*罕見的機柜是II級A2型生物平安機柜,主要區別在于它們的*小流入速度和排氣系統。 II類類型(右上)運用顏色說明在哪里可以平安地處置帶有微生物的風險化學物質。藍色是再循環空氣,在這里運用有害化學物質是不平安的。白色是單程空氣,關于化學運用是平安的。紫色表示由于生物平安柜煙霧位置的不確定性。圖表來源:維基媒體.圖表來源:微生物和生物醫學實驗室的生物平安(BMBL),第五版。附錄A 第三類也被稱為手套箱,提供*限制的維護;外殼是氣密的,一切資料經過浸泡罐或雙門高壓釜進出。III類柜用于高危微生物病原體或有毒化學品。 屏障隔離器:限制進入屏障系統(RABS),隔離器,微型環境 屏障隔離器是一個通用術語,包括兩種類型的設備:隔離器和限制進入屏障系統(RABS)。兩者都是在外部潔凈室環境和任務進程之間提供物理和空氣動力學(空氣過壓)屏障的裝置。 受限進入屏障系統(RABS) 受限通道護欄系統(RABS)便于任務和清潔。氣流從高效過濾器單向流出,自上而下流過關鍵任務區域。RAB的墻是實心的(玻璃或塑料片),RAB可以經過一扇門進入。在消費進程中,經過門、袖子/手套或半套裝進入該區域,*限制地增加人員與產品的接觸。RABS必需在潔凈的制藥區域停止消毒或滅菌。 隔離器和微型環境 隔離器是污染源和進程、產品或人員之間的屏障。它們主要用于制藥行業,以維護人員免受有毒化合物的污染。隔離器可以相關于它們所在的潔凈室被正壓或負壓。 產品的清潔度經過運用正壓隔離器來堅持,該隔離器將污染物從正在加工的資料中推開。負壓系統可用于容納風險資料,由于這將確保任何資料都不會超越相鄰的正壓邊界。 “微環境”相似于隔離器,但該術語通常用于半導體或納米技術的制造,而不是藥品。 在RABS與隔離器這兩者中,隔離器提供了更片面的處置環境與周圍設備的隔離。 潔凈室和分別設備的認證
實驗室需求確保它們的潔凈室和櫥柜經過認證,以確保它們契合所運用的規范。關于制藥和復合運用,NSF/ANSI規范49要求生物平安柜每年停止一次認證,或許在移動/重新放置生物平安柜時停止認證,由于HEPA過濾器能夠會在重新放置進程中移位。 與用于認證潔凈室的測試相似,關于生物平安柜等分別設備的認證測試:HEPA過濾器遭到粒子的應戰,浸透水平被測量以確保過濾器的完整性,一切氣流形式和流速被反省和調整以確保它們契合制造商的參數 規范 · ISO規范14644 *局部:依據顆粒濃度對空氣潔凈度停止分類 第二局部:經過顆粒濃度監測提供與空氣潔凈度相關的潔凈室功用證據 第三局部:測試方法 第七局部:分別設備(潔凈空氣罩、手套箱、隔離器和小環境) ISO 14698,潔凈室和相關受控環境—生物污染控制: *局部:普通準繩和方法 第二局部:生物污染數據的評價和解釋 歐盟GMP指南附錄1:無菌藥品的消費 美國美國食品藥品監視管理局公司(FDA)工業指南:經過無菌工藝消費的無菌藥品。 NSF/ANSI 49-2019:生物平安柜的設計和功用 潔凈室概述 潔凈室簡直運用于每一個小顆粒會抵消費進程發生不利影響的行業。潔凈室普遍用于半導體制造、制藥、生物技術、醫療設備和生命迷信等行業,以及航空航天、光學、軍事、核能和核醫先消費中罕見的關鍵工藝制造。 潔凈室是設計用來增加空氣中微粒污染和控制環境參數如溫度、濕度和壓力的任何封鎖空間。去除空氣中微粒的關鍵局部是用高效微粒空氣(HEPA)過濾器清潔空氣,或許在需求更嚴厲的清潔規范的中央,運用超低微粒空氣(ULPA)過濾器。假設任務環境需求過濾有毒氣體和氣息,可以運用高效氣體吸附過濾器(HEGA)去除空氣中的分子污染物。 污染與潔凈室分類的關系 潔凈室的設計、建造和運轉契合潔凈度分類的行業規范。房間所需的清潔度規范取決于在其中執行的義務;產品越容易遭到污染,所需的清潔規范就越高。 潔凈室是如何任務的? 潔凈室正常運轉必需滿足4個基本準繩: 1. 必需有足夠的量以每小時換氣次數來權衡的空氣流量,以稀釋或去除室內發生的污染物。 2. 潔凈室區域之間必需有足夠的壓差,以將空氣中的顆粒從潔凈區域推到不太潔凈的區域 3. 這質量提供應潔凈室的空氣不會給室內添加少量的污染 4. 潔凈室中的空氣活動應確保室內沒有高污染濃度的區域 假設滿足這些準繩,則應測量顆粒的濃度(在某些運用中,確定這些顆粒能否包括活的致病生物),以確保到達規則的潔凈室規范。然后,在一個繼續的基礎上,房間必需堅持契合規格: · 清潔 · 壓力 · 每小時換氣次數 · 溫度 · 濕度 · 流量 有些規范還要求測試光線和振動。 潔凈室分類 潔凈室依據空氣中微粒的允許濃度停止分類。有幾個樹立潔凈室分類的規范。 采用哪種規范由行業決議,*盛行的規范是ISO 14644,關于制藥和復合行業,還有歐盟良好消費規范(GMP)附錄1和FDA的GMP指南。 從ISO 1級到ISO 9級,ISO 1級被列為*潔凈的等級。ISO等級越低,環境越清潔。GMP將潔凈室分為A級到D級,A級大致相當于ISO 5級,D級相當于ISO 8級潔凈室 正壓和負壓潔凈室 大少數潔凈室都是正壓的,設計用于防止污染物進入室內。HEPA過濾的空氣通常從天花板水平引入潔凈室,其速率發生的空氣壓力大于室外的壓力。然后,空氣和空氣中的污染物被推向地板,并*終經過房間下部的通風口排出。這意味著空氣中的污染物不時流出房間。此外,經過通風口或當門翻開時排出房間的空氣具有足夠的壓力,以防止污染物經過這些啟齒進入。 邪氣壓潔凈室*常用于堅持污染物(如灰塵或碎屑)遠離潔凈室的運用中。例如,在制造進程中,它可用于維護醫院中的免疫缺陷患者或微芯片等敏感產品。 相比之下,負壓室的設計是為了防止污染物分開房間。在負壓室中,誤差經過反向HEPA過濾器從外殼中排出,在室內發生負壓,防止污染物分開房間,同時空氣經過通風口和其他啟齒不時被吸入。進入房間的空氣力防止污染物逸出。 賭氣壓潔凈室用于生化、制藥或醫療行業,尤其是在傳染病的狀況下。負壓室也用于化學品、易燃物和潛在爆炸性液體和粉末的一些風險處置。 氣流形式和通風 潔凈室曾經開展成兩種主要類型,它們的區別在于它們的通風方法。這些是紊流通風和直流潔凈室。紊流通風潔凈室也被稱為“非方向性的”。單向活動潔凈室*后被稱為“層流”潔凈室。單向型潔凈室比紊流通風型潔凈室運用更多的空氣,具有更高的潔凈度。 非單向氣流潔凈室有時用于ISO 6級潔凈室,但通常用于ISO 7級或更初級別。當堅持較低的空氣中顆粒濃度很重要時,在ISO級以下運用單向氣流。這兩種潔凈室如下圖所示。 圖1示出了一個湍流通風的房間,經過天花板中分散的空氣接納清潔的過濾空氣。這些空氣與室內空氣混合,并經過墻壁底部的抽氣裝置去除空氣中的污染物。換氣次數通常等于或大于每小時20次,這比普通房間如辦公室中運用的換氣次數大得多。在這種類型的潔凈室中,由人和機器發生的污染物與送風混兼并稀釋,然后被去除。
圖二。單向活動潔凈室。圖表來源:航天工業有限公司 圖2顯示了單向活動室的基本原理。高效過濾器裝置在整個天花板上(或某些系統中的墻壁上),這些過濾器提供空氣。這種空氣以單向方式掃過房間,并經過地板排出,從房間中去除空氣傳達的污染物。該系統比紊流通風潔凈室運用更多的空氣,但由于空氣的定向活動,它使室內的污染分散*小化。 潔凈空氣裝置,如單向任務臺或隔離器,用于紊流和單向通風潔凈室。這些機器將提供局部的過濾空氣,并在需求時改善空氣條件,例如在產品易受污染的區域。 進袋/出袋(碧波)赫加-HEPA過濾 在運用中表露在灰塵、煙霧或氣體中對維修技術人員來說是致命的,進袋/出袋(BIBO)過濾系統允許將用過的空氣過濾器放入渣滓袋。當需求改換過濾器時,過濾器檢修門被移除,顯露固定在門環上的縫焊袋。依照精心設計的順序移除用過的過濾器,然后改換過濾器,假設操作妥當,一切操作都不會表露過濾器外殼內的污染物。 碧波過濾系統運用效率極高的過濾器,如HEPA(高效微粒空氣)、ULPA(超低浸透空氣)或HEGA(高效氣相空氣)濾芯。大少數BIBO過濾器系統設計為多級系統:預過濾器級,接著是HEPA/ULPA級,能夠還有HEGA過濾器(假設氣流中能夠有氣體化合物或放射性同位素)。 BIBO系統在核工業中用于容納放射性同位素(發電;用于消費核醫學的盤旋減速器);觸及高效能或活性成分制造和包裝等的制藥運用。;運用通風櫥去除重金屬等有毒氣體的消費進程。;和醫院空氣隔離室(AIR's ),在那里空氣在沒有能夠的再夾帶的狀況下不能被排出。
美國醫療器械行業為何開展的這么好?

隨著中國經濟開展,越來越多的中國公司和投資人末尾關注海外醫療器械市場和投資。而美國作為擁有全世界*醫療器械市場的國度,成為關注的焦點。
首先,美國有弱小的消費力,2015年美國醫療器械市場占全球的40%,約為1400億美元。此外,美國也是醫療器械制造大國,2015年出口額達430億美元。其出色的消費和制造才干是吸引廣闊投資者來美國投資項目的重要緣由。
但是,明天我要在這里講講除了市場數據外,關于美國醫療器械投資,投資者們還需求知道的那些數據面前的故事。


美國醫療器械的創新環境
美國擁有世界上絕大少數*醫療器械公司,像百特醫療(Baxter),貝克曼庫爾特(Beckman Coulter),碧迪醫療器械(Becton Dickinson,*近收買了以醫用導管知名的巴德醫療,C.R. Bard Inc.),波士頓迷信(Boston Scientific), 通用醫療集團(GE Healthcare Technologies), 強生醫療(Johnson & Johnson),美敦力(Medtronic),史塞克公司(Stryker Corporation),康德樂(Cardinal Health,*近從美敦力收買了柯惠醫療,Covidien,的局部業務)等。
這些大公司,不只雇用了各種研發人才,也在孜孜不倦地支持著群眾創新。
強生的孵化器項目
強生孵化器項目借助強生公司現有的資源,為創業者和公司提供了一個更有效、更靈敏的創業平臺,減速新技術的商業化。每個進駐JLABS的創業公司,都可以享用強生公司從產品設計、法規咨詢,到資金和消費等各方面協助。
這種多方位支持,相比于傳統的天使投資或許風投,對很多初創公司更有吸引力。至今,已有312家優秀的創業公司入駐。其中有121名創業者曾是強生員工,JLABS吸納了這些有創意的前員工,為他們提供創業的環境和指點,同時又將這些項目牢牢抓在自己手里,這確實是一個高招。
JLABS
除了*的醫療器械公司,美國的高校在創新上也毫不示弱。一方面與大公司協作保送人才,一方面也開啟自己的孵化器項目。
高校的孵化器項目為學校里教職工和先生提供了積極友善的創業環境。波士頓和加州地域是美國著名的兩個醫療器械集聚區,那里有*高校,也云集了多所醫療器械公司和知名醫學院。
醫療器械產業第二興旺地域就是明尼蘇達州雙子城,她擁有全世界*的醫療器械公司,美敦力。此外,波士頓迷信,捷邁邦美(Zimmer Biomet),圣猶太醫療(St. Jude Medical)等公司也都在此設有分部。這里的明尼蘇達大學,專門成立了醫療器械研發中心(Earl E. Bakken Medical Devices Center)來支持創新項目;研發中心的項目跟明尼蘇達大學的科研嚴密相連,大學里豐厚的科研資金,也源源不時地為醫療器械研發中心提供新的創意。
明尼蘇達大學
更可貴的是,在距離雙子城一個半小時車程的羅切斯特城,有全世界知名的梅奧醫學中心(Mayo Clinic,全美排名*的醫院);明尼蘇達大學與梅奧醫學中心臨時展開醫學交流與協作。公司,高校和*醫院的結合,為醫療器械的創新提供了完美的環境,也是創新器械出現的基礎。
梅奧醫學中心
在美國的中西部還有一個正在逐漸崛起的醫療器械創新城市,猶他州的鹽湖城。
器械行業內比擬熟習的是這里的尼爾森實驗室(Nelson Lab于2016年被施潔醫療技術Sterigenics收買)。尼爾森實驗室有才干依照各項國際(ISO)和美國規范(ASTM,AAMI等),以及美國藥監局(FDA)的GLP要求,完成醫療器械的專項測試。這些測試結果被用于器械的法規注冊和產品聲明。尼爾森在提供測試的同時,也提供十分專業的咨詢效勞。

尼爾森實驗室
鹽湖城沒有特別大的醫療器械公司,但這里遍地是小規模的醫療器械創業公司,以開發和消費一次性器械為主。同時,這里也聚集了十分生動的天使投資人。我跟這里很多創業公司和創業人協作過,協助他們開發和消費醫療器械。隨著協作的深化,會逐漸提供設計咨詢、法規咨詢等。
美國醫療器械創新和創業的特點
創新,可以是一個從無到有的進程,也可以是一個改良和提高的進程。在我看來,美國醫療器械的創新,大致可以分為這么幾類。
改造型創新器械
是在功用上有打破性創新的醫療器械。比如目前越來越熱的手術機器人,其中*著名的要屬Intuitive surgical公司開發的達芬奇手術機器人。手術機器人通常用在微創手術中,可以大幅提高手術中器械操作的靈敏性,協助醫生在狹小空間完成復雜舉措,降低醫生的操作失誤,同時為完成更復雜的手術提供能夠。
有些復雜的手術,比如胸外科手術,由于對手術區域可視化的要求,采用傳統的器械,很難在微創(1~2cm)的條件下完成手術。傳統的方法是心內直視手術,來保證醫生有一個明晰的視野。但達芬奇手術機器人有高精度的攝像頭和精細控制的機械手,能輕松完成微創條件下的復雜手術。
達芬奇手術機器人
這類創新器械占的比例往往比擬小。他們對產品的設計和制造有十分高的要求,同時對FDA的審批人員也是一個應戰。由于FDA之前沒有接觸到過相似的器械,他們的同意進程其實變成了FDA自己一個學習和研討的進程。對企業來講,結果就是更嚴厲的要求和更長的審批時間。
改良型創新器械
大局部醫療器械在投入市場的時分,都不是盡善盡美的。醫學是一門系統性迷信,在治療中有太多的要素需求思索。即使是FDA在評價新器械的時分,也只是要求器械的效率大于風險(benefit over risk)。這就給醫療器械繼續的創新提供了條件。
一個很好的例子就是3D打印技術和新資料技術在植入物上的運用。3D打印為制造特性化假體提供了能夠。特性化的假體較比傳統的批量消費假體,有更多的優勢。它們取得病人生物數據,生成貼合病人生物學和力學結構的假體,延長了實踐手術時間,也防止了植入假體跟傷口不婚配的能夠。
FDA在2013年同意了*個3D打印的植入物,即OPM公司的3D打印聚合物顱骨假體。在2017年,另外一個美國公司SI-BONE的3D打印鈦金屬假體取得FDA同意。從聚合物到金屬的3D打印,是一個龐大的飛躍,由于金屬假體對資料的力學功用要求更高。
思索到目前3D打印受資料的局限性依然很大,新資料在這一類醫療器械方向的開展會有很大空間。未來新資料的崛起,比如可吸收資料,亦或力學功用與人體更接近的資料,還有增加免疫排擠的資料等,都將對3D打印假體的市場發生龐大的影響。
還有一種罕見的改良器械是對醫院本錢的優化。近期,在Biotochina路演上展現了一款由EDGe Surgical推出的一次性數字深度計。依據其預期用途,這一個新器械能替代目前脊柱融合手術中運用的7個器械。即使EDGe Surgical向美國醫院收取原來7個器械加起來一半的價錢來銷售這一個新器械,也將為醫院節省50%的本錢,醫院會更容易接受這個變卦。
法規在美國是一個不得不思索的方面。我們經常會聽說美國醫療范圍中的巨額賠償案例。例如2014年,紐約市急救中心的救護車上,由于沒有裝備先進的生命維持設備招致一名12歲患者嚴重的腦部損傷致使癱瘓,法院判決急救中心賠償患者1.72億美元。再如2007年,佛羅里達坦帕市醫院的醫務人員,由于對病人術后的感染控制不當,招致該病人失掉一切手指和雙腿的大局部,醫院被勒令賠償患者3千萬美元。
嚴厲的法規要求和巨額的賠償,促生了美國一大批防護性的醫療器械。特別是在感染防護(infection control),銳器損傷(sharps injury)等方面。美國疾病防控中心(CDC)在2009年發布統計,每年全美醫院由于病人在醫院中發作感染而招致的直接醫療費用為350億至450億美元,這里不包括醫院和保險公司前期支付給病人的賠償。
以此為商業契機,很多公司研發相應的器械,協助醫院改良環境,控制感染。例如Ivera Medical Corp(在2015年被3M公司以約1.53億美元收買),設計了一款用在滯留導管和輸液管上的防感染維護帽(Curos disinfecting port protectors)。當病人在輸完液后,護士就會為滯留在病人身上的導管蓋上這個一次性的維護帽(維護帽自身是滅菌的,外部還自帶酒精消毒),防止病人在活動時環境對導管的污染。
可以看出,美國不光有先進的技術和創新,社會制度和環境特點也反映到了其醫療器械的創新上。我們經常說醫療器械的設計不同于普通的消費品,需求思索到醫生、護士、病人等多方面用戶。同時,醫療器械的支付也不同于普通的消費品,很多時分運用者(醫生或許病人)并不是醫療器械的實踐支付者(經常由保險來支付,也能夠是醫院來支付)。這些都是我們在評價一個創新醫療器械的時分需求思索的。
美國醫療器械創業公司的特點
美國醫療器械的創業公司,跟很多其他創業公司一樣,往往末尾的時分只要幾團體。他們有共同的想法,設計了一款醫療器械,并完成了專利的注冊。假設是一款消費類產品,那么接上去,在拿到足夠的資本后,就可以消費和銷售了。
但是醫療器械不一樣,由于在法規上,它遭到美國FDA(U.S. Food and Drug Administration)的監管;在銷售上,很多醫院和診所都組織或許參與了GPO(Group Purchasing Organization),經過整合器械的運用量,跟器械商談判,來取得有利的推銷價錢。
不同于中國藥監局(CFDA),我以為美國FDA的法規對創新公司更為有利。作為一個初創公司,簡直不能夠有自己的工廠來消費產品。在消費范圍,*罕見的做法是創業公司把產品外包給第三方公司做代加工。
美國FDA也接受這個概念。只需產品設計公司(這個狀況下即創業公司)和代加工公司均在FDA注冊(備案),依照FDA的法規完成產品設計和消費即可。在這種狀況下,創業公司擔任醫療器械在FDA的法規注冊,從而擁有產品在FDA的注冊權,并可以隨時改換代工廠商。
但在目前CFDA的法規下,這一操作是不合規的。CFDA要求醫療器械的注冊商和制造商必需是同一家,而且目前實行的是審批制。這是目前一個影響國際醫療器械創新的中央。
除了設計和消費,醫療器械創業公司還少不了法規人員和銷售人員。
經過多年的開展,美國出現一大批合同制的法規人員和銷售人員(美國公司正式員工的雇用不存在合同,叫做at-will employment;只要暫時工或許短期工才是合同制,不享有公司福利。這與中國的勞工機制不同)。
關于新器械的開發和注冊(假設不是三類器械),法規人員的參與是有限的。這些擔任法規的人,往往以合同工或許咨詢(contractor/consultant)的方式,協助創業公司完成新器械的FDA注冊,并在需求時提供相應的法規咨詢。
器械的銷售人員也相似,他們跟左近的醫院和診所十分熟習,為各個醫療器械公司提供銷售效勞,賺取傭金。這些人員我稱之為基礎設備,他們的普遍存在,很大水平上延長了器械創新和上市的周期。
美國創新器械進入中國市場的討論
前面我們說到,美國是目前全世界醫療器械消費*的市場,而中國,則是醫療器械市場開展*快的國度。20120.1%,其中72.7%的增長來自醫院的推銷。
隨著人們生活水平逐漸提高,城鄉化差距的增加,以及老齡化的減速,中國對醫療技術和產品的追求會不時提高。包括*近愈演愈烈的赴美就醫,赴日就醫,都是國際日益增長的醫療需求得不到滿足的表現。
同時,我們知道全球第七大醫療公司康德樂在2017年11月正式出售了其在中國的全部醫藥業務,但依然保管了在華醫療器械業務。可以看出國際公司對中國醫療器械行業開展的看好。
將美國創新醫療器械引入中國的時分,除了準確預估市場大小,還需求從以下幾個方面思索。
支付方式
醫療器械很多時分不是由用戶(患者)來直接支付的,觸及到保險公司、醫院等等。即使用戶的需求在,但假設中美的支付方式不一樣,*后構成的市場大小也會有差異。
法規評價
除了前面提到的,CFDA對國產器械注冊商和制造商的捆綁要求之外,當國外*新技術進入中國市場時,需求思索CFDA對新技術的接受水平。
從歷史閱歷來看,CFDA無論從法規要求的制定還是對新器械的同意,都或多或少在效仿和跟隨FDA的步伐。在對新技術和器械的同意上,很難逾越FDA。即使FDA曾經同意了的器械,假設技術上是超前的,CFDA也需求一段時間來了解和順應。這就會影響產品上市的時間。
市場接受
前面提到,美國由于其縝密的法規要求和巨額的懲罰性賠償,培育了一些市場特有的醫療器械。這些器械,在其他市場并不一定適用。
一個很好的例子是,美國普遍運用的一次性器械的概念。從各式各樣的一次性無菌器械,到各大醫院手術室運用的特性化手術包概念,根絕了器械由于重復滅菌運用帶來的感染和器械功用下降的風險。
相比擬,中國手術室目前較多還在用重復運用的器械。由于重復滅菌的本錢和其他風險本錢缺乏以讓中國醫院去接受一次性器械的價錢。
厚著臉皮給自己打個廣告。歡迎大家來我的淘寶店鋪“青島口罩網”轉轉。新店鉅惠停止中,84消毒液僅售0.9元,N95口罩、兒童口罩等等全部底價現貨銷售,還有全場通用的滿減活動!歡迎進入“青島口罩網”淘寶店鋪選購。
更多醫療器械行業資訊,請關注我們的群眾號:凱象。凱象平臺,一個專為醫療器械消費、銷售企業打造的全球尋源平臺。我們將為您打通時間、空間的壁壘,為供需雙方樹立一座直接交流的橋梁。讓您足不出戶找產品,無需見面談生意!
珠海防護用品消毒劑檢測?美國醫療器械行業為何開展的這么好? 健明迪檢測
《北京市醫療器械審評核對咨訊問答300問》(中冊)
*篇 有源產品
101. 第二類有源醫療器械產品央求變卦注冊添加型號,什么狀況下可以由原有型號的檢測報告掩蓋,什么狀況下必需停止檢測?
答:首先應確認所央求添加的型號與原有型號能否可作為同一注冊單元,如可作為同一注冊單元,可央求變卦注冊添加型號。在不觸及新規范的狀況下,應當依照典型性型號的判定準繩,如原有型號可代表新增型號,則無需重復停止檢測;如觸及新規范,則需提供新增型號針對新規范的檢測報告;如原有型號的檢測報告中局部項目檢測可代表新增型號檢測,則此局部外容無需重復檢測。
102.關于管理類別由第三類調整為第二類的醫療器械產品,其產品注冊證還為第三類產品注冊證,假設產品發作了《醫療器械注冊與備案管理方法》中規則的需停止變卦注冊的變化,能否直接到住所所在的省、自治區、直轄市藥品監視管理部門央求操持變卦注冊?
答:此種狀況,不能直接到住所所在的省、自治區、直轄市藥品監視管理部門央求操持變卦注冊,應到原注冊部門央求操持變卦注冊手續。
103.第二類有源醫療器械產品的產品技術要求中,“產品型號/規格及其劃分說明”在撰寫進程中應留意哪些效果?
答:依據《醫療器械產品技術要求編寫指點準繩》,產品技術要求中應明白產品型號和/或規格,以及其劃分的說明。產品型號/規格及其劃分說明中普通應明白產品的結構及組成,對同一注冊單元中存在多種型號和/或規格的產品,應明白各型號及各規格之間的一切區別,必要時可附相應圖示停止說明。關于型號/規格的表述文本較大的可以附錄方式提供。
關于獨立軟件或含有軟件組件的產品,還應明白軟件的稱號、型號規格、發布版本、完整版本的命名規則,控制型軟件組件還應明白運轉環境(包括硬件配置、軟件環境和網絡條件)。
104.《醫療器械網絡平安注冊審查指點準繩》適用于哪些醫療器械?
答:《醫療器械網絡平安注冊審查指點準繩》適用于醫療器械網絡平安的注冊申報,包括具有電子數據交流、遠程訪問與控制、用戶訪問三種功用當中一種及以上功用的第二、三類獨立軟件和含有軟件組件的醫療器械(包括體外診斷醫療器械);適用于自研軟件、現成軟件的注冊申報。
其中,網絡包括無線、有線網絡,電子數據交流包括基于網絡、存儲媒介的單向、雙向數據傳輸,遠程訪問與控制包括基于網絡的實時、非實時的訪問與控制,用戶(如醫務人員、患者、維護人員等)訪問包括基于軟件用戶界面、電子接口的人機交互方式。
105.關于產品技術要求不觸及或局部觸及國度規范、行業規范停止檢驗并出具報告的,沒有CMA章,如何證明報告合規?
答:依據《國度藥監局綜合司關于明白〈醫療器械檢驗任務規范〉標注資質認定標志有關事項的通知》(藥監綜科外函〔2020〕746號)的規則:關于產品技術要求不觸及或局部觸及國度規范、行業規范停止檢驗并出具報告的,應在檢驗報告書備注中對承檢才干予以自我聲明,并承當相應的法律責任。自我聲明內容為“該產品技術要求不觸及/局部觸及國度規范、行業規范,不能直接作為資質認定容許的依據,但本實驗室對報告觸及的檢驗項目具有相應的承檢才干”。
106.第二類有源醫療器械產品在停止電磁兼容檢測時,能否需求連同產品組成中的無源附件一同檢測?
答:通常電磁兼容檢驗中運用的設備裝置、電纜布局和典型配置中的全部附件應與正常運用時分歧。假設經剖析判定無源附件與電磁兼容檢驗有關,則不需求連同該無源附件一同檢測。假設測試時必需配合無源附件才干完成其基本功用,則應當配合該無源附件停止檢測。
107.含軟件的第二類有源醫療器械產品在用同種類比對的方式停止臨床評價時,關于軟件差異應如何思索?
答:對比時,注冊央求人應詳細描畫軟件相關的一切差異,剖析差異能否對產品的平安性、有效性發生的影響。必要時,應提交申報產品自身的臨床/非臨床數據來證明該差異未對平安有效性發生不利影響。
108.第二類有源醫療器械產品中包括多個模塊,每個獨自的模塊都是《免于臨床評價醫療器械目錄》中的產品,組合產品能否也可視為《目錄》中產品?
答:假設央求人可以證明兩者的組合不存在相互影響,且臨床用途未超出豁免目錄范圍,則可以為是兩種豁免產品的復雜組合,仍按《目錄》中產品臨床評價要求對兩個模塊區分停止評價,央求人須評價模塊組合能夠帶來的風險。
109.《醫療器械網絡平安注冊審查指點準繩》只適用于網絡平安的范圍嗎?能否思索信息平安和數據平安?
答:信息平安、網絡平安、數據平安的定義和范圍各有側重,既有聯絡又有區別,不盡相反,但該指點準繩從醫療器械平安有效性評價角度動身對三者不做嚴厲區分,一致采用網絡平安停止表述,即從網絡平安角度綜合思索醫療器械的信息平安和數據平安。
110.電磁兼容檢測的抗擾度實驗中產品基本功用如何確定?
答:基本功用是指必需的功用,制造商在確定產品基本功用時,應思索但不限于以下方面:剖析臨床平安性風險,思索和診斷/治療/監護相關的功用,各種傳感器、線纜、運用局部、控制裝置、顯示裝置、運動部件等功用能否受電磁攪擾影響。
隨機文件所識別的基本功用應作為基本功用停止抗擾度實驗。假設未在隨機文件中識別出基本功用,全部功用均應思索作為基本功用停止抗擾度實驗。
111.如何對同種類醫療器械臨床數據停止搜集、評價和剖析?
答:同種類醫療器械臨床數據需合法獲取,包括臨床文獻數據、臨床閱歷數據和臨床實驗數據。為充沛識別申報產品臨床風險的種類和水平,準確表征其臨床功用、有效性、臨床收益和所處的行業水平,注冊央求人需依據《醫療器械臨床評價技術指點準繩》提出的準繩和要求,對同種類醫療器械臨床數據停止搜集、評價和剖析。
112.關于第二類有源醫療器械產品,產品結構組成中某個部件屬于非醫療器械,在產品注冊證書的結構組成中能否可以表現該部件?能否需求對該部件停止檢測?答:關于不作為醫療器械管理的產品部件,不能獨自申報注冊,但可以作為醫療器械的組成局部。
假設該部件作為醫療器械組成局部申報,那么應將其視為全體的一局部停止評價,該部件應隨零件一同停止相應的檢測、驗證。假設央求人未將該部件隨零件一同停止檢測、驗證,則不能同意其作為產品組成局部。例如:計算機、打印機、臺車、電纜線、內窺鏡照明光纜、中性電極銜接線等,都屬于此種情形。
113.第二類有源設備的開關方向是如何規則的?
答:GB9706.1中57.1章節要求第二類有源設備的電源總開關、控制機器的總電源開關,其方向應契合GB4205,上開下關,左關右開。其他開關,比如電腦下面的待機開關沒有該要求。
114.第二類有源醫療器械產品變卦注冊時電氣元件不發作變化,發作其他變化,如外殼結構改動、設備全體密封功用改動等,能否可以豁免電氣平安和電磁兼容檢測?
答:應對申報產品變化狀況停止全體評價,若外殼結構改動、密封功用改動等觸及電氣平安規范/電磁兼容規范中局部項目的變化,需重新判定或評價,則應停止檢測。
115.第二類有源醫療器械產品關鍵元器件供應商發作變化需求操持變卦注冊嗎?
答:假設關鍵元器件供應商在產品技術要求中曾經載明,供應商發作變卦則需求央求操持變卦注冊;假設沒有在產品技術要求中載明,可不停止變卦注冊,但應依照質量管理體系的要求停止控制,對相關設計更改停止風險剖析,必要時采取措施將風險降低到可接受水平。
116.配算計算機運用的第二類有源醫療器械產品,產品技術要求中需對計算機配置停止描畫,應如何描畫才干防止由于計算機配置變化頻繁招致變卦注冊頻繁停止?
答:產品技術要求中對計算機配置停止描畫時,如CPU頻率、存儲空間、內存空間、顯示器分辨率等,可描畫配置的*要求。計算機配置發作變化時,不觸及產品技術要求變化的,可不停止變卦注冊,但應依照質量管理體系的要求停止控制,對相關設計更改停止驗證。
117.第二類有源醫療器械產品顏色發作變化需求操持變卦注冊嗎?
答:假設注冊證及其附件中未載明產品顏色,則無需操持變卦注冊,企業應依照其自身質量管理體系要求做好相關任務。假設注冊證及其附件中載明了產品顏色,則需求操持變卦注冊。
118.第二類有源設備配合軟件運用,設備和軟件都有獨自的注冊證書,設備的注冊證書中明白了配合運用軟件的稱號和發布版本號。該軟件的發布版本晉級后已停止了變卦注冊,那么設備注冊證書中該軟件發布版天分否在延續的時分直接變卦為新版本?能否需求央求變卦注冊?
答:配合運用軟件的發布版本號為設備注冊證中載明事項,不能在延續注冊時停止變卦。假設設備要配合變卦后版本的軟件運用,應央求變卦注冊,在變卦注冊時對配合運用的平安、有效性停止論證;如未停止變卦,則設備不能配合新版本的軟件運用,但仍可繼續配合變卦前版本的軟件運用。
119.人工智能醫用軟件一定是第三類醫療器械嗎?
答:不是,關于算法在醫療運用中成熟度低(指未上市或平安有效性尚未失掉充沛證明)的人工智能醫用軟件,若用于輔佐決策,如提供病灶特征識別、病變性質判定、用藥指點、治療方案制定等臨床診療建議,依照第三類醫療器械管理;若用于非輔佐決策,如停止數據處置和測量等提供臨床參考信息,依照第二類醫療器械管理。
120.第二類有源醫療器械產品中可重復運用的附件消毒滅菌資料應關注的效果有哪些?
答:可重復運用的附件,運用前應保證已消毒或滅菌。說明書中應明白詳細的消毒/滅菌方法(如運用的消毒劑、消毒或滅菌設備),消毒/滅菌周期的重要參數(如時間、溫度和壓力等)。研討資料中應提供消毒/滅菌方法確定的依據、消毒/滅菌效果確認資料及引薦的消毒/滅菌方法耐受性的研討資料。
121.延續注冊后的新注冊證未失效,此時需求做變卦注冊或變卦備案,應運用哪個注冊證停止變卦?
答:應運用在注冊證有效期內的注冊證停止變卦。
122.有源產品申報延續注冊時,應如何撰寫“產品概述”和“有關產品平安性、有效性主要評價內容”?
答:“產品概述”中建議論述:任務原理或作用機理;適用范圍;結構組成;注冊證號及有效期。
“有關產品平安性、有效性主要評價內容”中建議論述:
(1)本次延續注冊進程中該產品無變化;
(2)若本次注冊證有效期內產品變卦狀況,應明白變卦時間和變卦事項;
(3)若本次醫療器械注冊證有效期內有新的醫療器械強迫性規范發布實施,為契合新的醫療器械強迫性規范所做變卦狀況;
(4)若本次延續注冊為注冊管理類別有調整的產品,應注明管理類別調整文件,并明白調整前管理類別和調整后管理類別;
(5)若本次延續注冊為分類編碼有調整的產品,應注明分類編碼調整文件,并明白調整前分類編碼和調整后分類編碼;
(6)若原醫療器械注冊證中載明要求繼續完成的事項,應說明事項的完成狀況;
(7)總結陳說本次延續注冊產品平安有效性,能否契合延續注冊要求。
第二篇 無源產品
123.一次性運用腹部穿刺器技術要求中應包括哪些物理功用?
答:一次性運用腹部穿刺器技術要求中應包括 YY/T 1710《一次性運用腹部穿刺器》適用的相關功用,假設申 報產品的結構不同于 YY/T 1710 的設計結構,或許在 YY/ T1710給出的結構基礎上還有其它設計元素,應制定與之相關的功用要求,如以空氣密封壓力屏障設計的穿刺器的走漏率、穿刺桿透明*與腹腔鏡的配合功用(若適用)、固定裝置(若有)的功用、平安維護罩(若有)的功用、吸收液體的組件(若有)功用等。
124.一次性運用腹部穿刺器技術要求中應包括哪些化學功用?
答:包括但不限于:浸提液的濁度及色澤;恢復物質、重金屬含量、酸堿度、蒸發殘渣;環氧乙烷殘留量(如適用);若有涂層,需制定涂層有關功用;若含有吸收液體的組件,需制定吸收劑溶出相關的功用。
125.免臨床目錄產品如何提交評價資料?
答:依據《列入免于臨床評價醫療器械目錄產品對比說明技術指點準繩》,關于列入《免于臨床評價醫療器械目錄》(以下簡稱《目錄》)產品,注冊央求人需提交申報產品相關信息與《目錄》所述內容的對比資料和申報產品與已獲準境內注冊的《目錄》中醫療器械的對比說明。詳細需提交的資料要求如下:
(一)提交申報產品相關信息與《目錄》所述內容的對比資料;
(二)提交申報產品與《目錄》中已獲準境內注冊醫療器械的對比說明,對比說明應當包括《申報產品與目錄中已獲準境內注冊醫療器械對比表》和相應支持性資料。若經對比,申報產品與對比產品存在差異,還應提交差異局部對平安有效性影響的剖析研討資料。二者的差異不應惹起不同的平安有效性效果,即申報產品未出現對比產品不存在的且能夠引發嚴重風險和/或惹起清楚影響有效性的效果。
提交的上述資料應能證明申報產品與《目錄》所述的產品具有基本等異性。若無法證明申報產品與《目錄》所述的產品具有基本等異性,則應展開臨床評價。
126.脫敏劑能否需求在技術要求中明白產品組分?
答:建議在產品技術要求型號規格及劃分說明中列明各組分的稱號、含量,各組分比例之和應為100%。
127.如何劃分脫敏劑的注冊單元?
答:脫敏劑注冊單元準繩上以產品的技術原理、結構組成、功用目的和適用范圍為劃分依據,例如:產品作用機理不同,建議不作為同一注冊單元申報;產品主要成分不同,建議不作為同一注冊單元申報。
128.包皮切割吻合器如何選取檢驗典型型號?
答:包皮切割吻合器抽取樣品應能涵蓋該注冊單元全部產品的功用目的,組成資料、結構功用等具有差異的產品,建議區分選取典型性型號停止差異性檢驗。例如:吻合釘資料不同,如鈦與不銹鋼,應區分在同種資料的產品中選擇典型產品停止檢驗。
129.包皮切割吻合器吻合釘能否豁免生物學實驗?
答:包皮切割吻合器吻合釘目前多采用鈦、鈦合金或純鉭、不銹鋼資料。吻合釘普通應選用契合GB/T13810中化學成分要求的純鈦、鈦合金資料,或契合YY/T0245、ISO13782中化學成分要求的純鉭資料,或契合GB4234.1中化學成分要求的不銹鋼資料。關于采用以上資料制造的吻合釘可豁免生物學實驗,并將吻合釘成分證明資料作為生物相容性評價研討資料的一局部。選用其他資料(如其他不銹鋼資料、其他金屬資料)或外表改性處置的純鈦、鈦合金、純鉭等制成的吻合釘,應依照GB/T16886《醫療器械生物學評價》系列規范對吻合釘停止生物相容性評價研討,普通包括但不限于細胞毒性、致敏、皮內反響、急性毒性、亞慢性毒性、遺傳毒性和植入后局部反響。
130.關節鏡下無源手術器械如何命名?
答:關節鏡下無源手術器械的命名應契合相關法規、國度規范、行業規范的要求。關節鏡下無源手術器械通常由1個中心詞和不超越3個特征詞確定產品通用稱號,可按作用對象和預期用途等方式來命名,如膝關節鏡用手術抓鉗、膝關節半圓型銼、關節鏡下鳥嘴鉗等。關節鏡下無源手術器械包類產品以表現產品組成、功用用途為基本準繩,由多種類別器械組成,普通應以其主要預期用途來命名,如髖關節內窺鏡手術器械包。
131.如何劃分關節鏡下無源手術器械的注冊單元?
答:注冊單元劃分應依據相關法規文件要求,醫療器械注冊單元準繩上以產品的技術原理、結構組成、功用目的和適用范圍為劃分依據。關節鏡下無源手術器械產品不同規格、型號可為同一注冊單元。以無菌方式和非無菌方式提供的關節鏡下無源手術器械產品可劃為同一注冊單元。關節鏡下無源手術器械包注冊單元的劃分應首先思索預期用途,預期用途不同的手術包不作為同一單元。
132.如何劃分正畸絲的規格型號?
答:正畸絲可按預成型外形、運用部位組成股數、截面外形等不同分為若干型號:按預成型外形可分為直絲和預成型弓絲,其中預成型弓絲按預成型外形的特征尺寸(弓形高、弓形寬、中心半徑等)不同又可分為不同弓形;運用部位通常分為上頜和下頜;按組成股數可分為單股正畸絲和多股正畸絲,多股正畸絲由兩支或多支單絲,經過扭絞、編織或其他工藝制成;按截面外形可分為圓絲和方絲。II型正畸絲還可按奧氏體轉變完畢的溫度不同區分不同型號。正畸絲按截面尺寸的大小又可以分紅不同規格。截面尺寸可用單股圓絲截面的直徑正好容納多股圓絲的管子的內徑或方絲截面的寬度、高度和對角線表示。依照正畸絲慣例通常采用描畫符(即無單位稱號的千分之一英寸)作為識別代碼。
133.正畸絲產品技術要求應包括哪些功用目的?
答:正畸絲產品的功用目的和檢驗方法可參考相應行業規范YY/T 0625。若對規范中的功用目的或實驗方法有所修正,應說明修正的內容及緣由,并提交驗證資料。通常應思索(但不限于)以下功用目的:(1)外觀;(2)尺寸及公差要求;(3)金屬外表粗糙度;(4)耐腐蝕性;(5)奧氏體轉變完畢溫度(II型正畸絲適用);(6)力學功用;(7)資料的化學成分;(8)有害元素。若注冊央求人宣稱產品具有特定的技術特征(如微生物功用、涂層功用等),應制定相應的功用目的。
134.經環氧乙烷滅菌后無菌提供的第二類無源醫療器械,在申報初次注冊時應提供哪些滅菌驗證資料?
答:滅菌驗證資料應包括滅菌方案及滅菌報告,報告中至少應包括滅菌的主要信息(如滅菌批號,消費批號) 及滅菌結果、滅菌結論。滅菌結果應以列表的方式論述滅菌進程中的數據匯總信息(如短周期、半周期及全周期的實驗結果,菌片、IPCD、EPCD及產品的檢測結果);滅菌結論應包括滅菌參數和裝載圖,裝載圖應至少能表現滅菌驗證中菌片、溫度傳感器的放置狀況。滅菌驗證的原始記載,應提交各個周期的滅菌批記載單(包括滅菌批號、消費批號和委托滅菌企業公章)和滅菌效勞協議(應包括產品清單),其他原始記載依據滅菌驗證資料的完整性選擇提供或不提供。
135.第二類無源醫療器械申報初次注冊時,應提供的原資料材質證明有哪些要求?
答:除義齒類產品依照相關文件要求提供原資料注冊證信息外,其他產品應提供原資料材質單。材質單可以是原資料供方提供出廠檢驗報告、委托檢測報告或由注冊人委托檢測機構出具的原資料檢測報告。
136.第二類無源醫療器械申報初次注冊時,注冊人提供的綜述資料中,原資料局部應論述哪些信息?
答:針對原資料的相關信息,注冊人應論述與運用者和/或患者直接或直接接觸的資料成分;論述所包括的生物資料或衍生物的物質來源、原資料、預期運用目的、主要作用方式、應契合的材質規范、供方提供的材質證明資料等;論述從原資料到成品整個加工工藝進程中所運用的加工助劑、加工助劑發揚的作用、加工助劑的質控規范、加工助劑的去除、加工助劑*終在產品中的殘留方式/殘留量/平安限值說明等。
137.第二類無源醫療器械申報初次注冊時,注冊人應提供哪些資料停止生物相容性評價?
答:關于與患者直接或直接接觸的器械,應當停止生物學評價。生物學評價資料應當包括:
(1)描畫產品所用資料及與人體接觸性質,設計和消費進程中能夠引入的污染物和殘留物,設計和消費進程中能夠發生的析出物(包括濾瀝物和/或蒸發物)、降解產物、加工殘留物,與醫療器械直接接觸的包裝資料等相關信息;
(2)描畫申報產品的物理和/或化學信息并思索資料表征(如適用),如器械的物理作用能夠發生生物學風險,應當停止評價;
(3)生物學評價的戰略、依據和方法;
(4)已有數據和結果的評價;
(5)選擇或豁免生物學實驗的理由和論證;
(6)完成生物學評價所需的其他數據。
針對上述資料,注冊人應確保以下內容:生物學評價依據的規范應正確;評價的項目完全;提交的已有數據/結果應與申報產品可樹立起溯源關系,且有支持性證據。生物學報告應包括契合相應規范的實驗進程的詳細信息,并與申報產品可樹立起溯源關系;報告應提交原件。
138.第二類無源醫療器械申報初次注冊時在綜述資料中如何描畫產品研發歷程?
答:注冊人應論述申報注冊產品的研發背景和目的。如有參考的同類產品或前代產品,應當提供同類產品或前代產品的信息,并說明選擇其作為研發參考的緣由;除上述要求外,注冊人應論述申報產品所處置的臨床效果,處置該臨床效果的以后現有手腕、不同處置手腕的優缺陷比擬、開展趨向,與本申報產品任務原理/作用機理相反的同類產品市場狀況、主要品牌、各自的差異性、優缺陷,本申報產品研發參考的同類產品或前代產品,選擇其作為研發參考的緣由。
139.第二類無源醫療器械申報初次注冊時在綜述資料中如何描畫注冊產品與同類和/或前代產品的參考和比擬?
答:注冊人應論述注冊產品與同類和/或前代產品的差異性,可以以列表的方式對以下內容停止比擬并說明其差異性:產品命名、任務原理/作用機理、規格型號、結構組成、制造資料、制造工藝/加工助劑運用、功用目的、作用方式(如植入/介入)、適用范圍、忌諱證、留意事項、提供方式(如無菌提供/非無菌提供/微生物限制控制要求)、產品有效期、產品包裝、產品運用次數(如一次性運用/可重復運用)等。
140.定制式固定義齒如何選擇典型型號停止檢測?
答:依據《定制式義齒注冊技術審查指點準繩》,依照“同一注冊單元內,所檢測的產品應當是可以代表本注冊單元內全部產品平安性和有效性的典型產品”的準繩,典型產品是指可以涵蓋本注冊單元內全部產品工藝的一個或多個產品。如固定義齒可抽取數量不低于3單位的金屬烤瓷橋(鑄造、堆積等)、金屬橋(鑄造、堆積等)和全瓷橋(鑄瓷、CAD/CAM等)停止檢測。如企業只消費單冠產品,可抽取1顆單冠停止檢測。數量不低于3單位的橋可以掩蓋相反工藝的冠、貼面、樁核和嵌體。
141.第二類無源醫療器械申報初次注冊時,消費制造信息至少應論述哪些信息?
答:初次注冊資料中消費制造信息至少應論述總體消費工藝的簡明說明、消費工藝流程圖、關鍵工序控制說明、特殊進程控制參數、外協工序說明(含供應商稱號和地址)、消費檢驗設備清單、消費場地引見、研發場地引見、場地平面圖等。
142.第二類無源醫療器械申報注冊時,援用的通用規范和公用規范中功用要求有差異時,應該如何制定產品技術要求的功用目的?
答:首先應思索能否為強迫性規范,無論通用規范還是公用規范,強迫性規范級別*;假設為同級別,即都是強迫性規范或都是引薦性規范,通常狀況公用規范優先于通用規范。在上述基本準繩下,建議注冊央求人綜合思索兩個規范的功用要求。
143.第二類無源醫療器械申報延續注冊時,央求表中應如何撰寫“產品概述”和“有關產品平安性、有效性主要評價內容”?
答:“產品概述”中建議論述:任務原理或作用機理;適用范圍;結構組成;主要資料、能否無菌提供、能否一次性運用;注冊證號及有效期。
“有關產品平安性、有效性主要評價內容”中建議論述:
(1)本次延續注冊進程中該產品無變化;
(2)若本次醫療器械注冊證有效期內產品有變卦,應說明變卦時間和變卦事項;
(3)若本次醫療器械注冊證有效期內有新的醫療器械強迫性規范發布實施,應說明為契合新的醫療器械強迫性規范所做的變卦狀況;
(4)若本次延續注冊管理類別有調整,應注明管理類別調整文件,并說明調整前管理類別和調整后管理類別;
(5)若本次延續注冊分類編碼有調整,應注明分類編碼調整文件,并說明調整前分類編碼和調整后分類編碼;
(6)若原醫療器械注冊證中載明了要求繼續完成的事項,應說明事項的完成狀況;
(7)總結陳說本次延續注冊產品平安有效性,能否契合延續注冊要求。
144.延續注冊產品為契合新的強迫性規范發布實施所做的變化屬于無需操持變卦注冊手續或許無需變化的,應提供哪些申報資料?
答:(1)已注冊產品為契合新的強迫性規范發布實施所做的變化屬于無需操持變卦注冊手續或許無需變化的說明;
(2)若有無需操持變卦注冊手續的變化,應提供相關證明資料(至少包括新舊規范觸及條款原文復印件和新舊規范觸及條款對比表)。
第三篇 臨床檢驗產品
145.第二類體外診斷設備的產品技術要求中,環境實驗還要求嗎?
答:依照《醫療器械產品技術要求編寫指點準繩》相關肉體,環境實驗可不在產品技術要求中表現。但是相關研討還是應在產品研發進程中表現,并在注冊申報時提交相關研討資料。
146.說明書中主要組成成分應包括哪些?
答:應包括*小銷售單元中一切參與反響的組分(如各種反響試劑、檢測卡)以及檢驗進程中輔佐成分(如枯燥劑、比色卡、封板膜、生物平安袋、滴管、校準信息碼等)。不含說明書。
147.試劑盒組分獨自銷售時,應留意什么?
答:銷售包裝標簽應包括完整試劑盒相關信息(如產品稱號、注冊人、消費地址等),并應注明組分基本信息(包裝規格、貯存條件及有效期、批號等)以及需留意事項。包裝應附原試劑盒說明書。如觸及UDI賦碼,應契合相關法規要求。
148.半定量產品的準確度如何要求?
答:可將準確度、檢測限兼并要求,關于每個分段點均應驗證。*要求為:檢測結果與相應參考溶液標示值相差同向不超越一個量級,不得出現反向相差,陽性參考溶液不得出現陰性結果,陰性參考溶液不得出現陽性結果。各量級檢測應到達95%的契合率。
149.產品技術要求中,特異性如何要求?
答:產品技術要求中的特異性,普通是指交叉反響,而非攪擾。所以普通關于非免疫比濁法的生化試劑無此要求,免疫類產品如有明白的交叉反響物(如不同亞型等)應有此要求。添加的交叉反響物濃度應選擇臨床上可見的較高水平。
150.瓶間差何時要求?
答:干粉試劑,普通應有瓶間差及復溶動搖性的要求。
151.臨床評價時,關于多種樣本類型,如何要求?
答:假設申報的試劑產品同時適用于血清、血漿和/或全血樣本,那么樣本散布應為:血清和血漿可以為屬于同一類型停止總量統計(但是需求前期剖析功用評價驗證兩者分歧性);如既有血清/血漿,又有全血樣本,則應停止同源樣本比對(例數按法規要求),且全血正常、異常值的散布要求同血清/血漿。但是血液、尿液、腦脊液等樣本之間不適用此狀況,應區分停止臨床評價。
152.體外診斷試劑包裝、標簽的設計作風變化、說明書排版和格式的調整,能否需求停止變卦注冊?
答:不需求。IVD說明書中其他可自行變卦的內容參見《總局辦公廳關于體外診斷試劑說明書文字性變卦有關效果的通知》(食藥監辦械管〔2016〕117號)。
153.局部腫瘤標志物由三類降為第二類后,說明書中“預期用途”描畫如何修正?
答:腫標降類后,央求表中的“預期用途”的寫法與慣例產品堅持分歧。說明書中【預期用途】*段應與央求表中堅持分歧(同第二類慣例產品要求),第二段應以降類文件中調整后的預期用途“臨床上用于……”掃尾,并添加“不用于普通人群的腫瘤篩查”的表述。第二段后續內容與慣例產品堅持分歧,描畫相關臨床意義即可。
154.央求表中,“預期用途”如何描畫?
答:試劑盒應明白“體外”“定性/定量”“樣本類型”“被測物”等信息,校準質控品需明白一切項目。舉例如下:
例1:用于體外定性檢測人血清、血漿中××××的活性。
例2:用于體外定量測定人血清中×××的含量。
例3:與本公司的試劑盒配套運用,用于××檢測系統的校準。
例4:與本公司的試劑盒配套運用,用于A、B、C……共×項的室內質量控制。
155.關于超敏C反響蛋白試劑盒,其預期用途中被測物能否為“超敏C反響蛋白”?
答:這種了解是錯誤的。被測物依然為“C反響蛋白”,“超敏”僅是關于該類產品靈敏度高的一種表述。所以應表述為“用于體外定量檢測人血清、血漿中C反響蛋白的含量”。
156.產品按定量還是定性停止申報,需求思索哪些要素?
答:首先應該思索臨床實踐運用需求以及能否有同類產品已上市,其次樣本類型也應該思索,普通關于大便、唾液、咽拭子等非均質化樣本很難真正意義上完成“定量”檢測。
157.產品想申報家用或患者自測,應該思索哪些要素?
答:首先應思索該產品能否有家用或患者自測需求及運用價值,其次應思索樣本取得的方便性,普通應為尿液、唾液、指尖血等。詳細要求可參閱《家用體外診斷醫療器械注冊技術審查指點準繩》。
158.說明書中產品英文稱號有何要求?
答:不建議寫英文稱號,如確需必要,英文稱號應當正確、完整,不允許只寫縮寫。應直譯,而非意譯。
159.關于產品稱號中方法學局部,有何要求?
答:生化試劑盒可參考YY/T1227-2014《臨床化學體外診斷試劑(盒)命名》,不建議運用的方法學表述:終點法、速率法、延續監測法、IFCC法等。膠體金法應為“膠體金免疫層析法”,化學發光法應為“化學發光免疫剖析法”或“磁微粒化學發光免疫剖析法”。
160.產品包裝規格如何規范表述?
答:不同包裝規格之間應依照分隔層次區分運用頓號、逗號、分號停止區分,一致以句號完畢。如僅單一包裝規格,其后可以不加標點符號。舉例如下:
例1:48人份/盒
例2:48測試/盒、96 測試 / 盒。
例 3: 試劑 1(R1):5mL×5, 試劑 2(R2):5mL×5; 試劑 1(R1):10mL×10,試劑 2(R2):10mL×10。
例 4:反響液:15mL×1, 緩沖液:45mL×1, 校準品: 2mL×1。
例 5: ××× 型:4mL×10(凍干品,復溶體積)
161.產品技術要求中,線性如何要求?
答:留意區間的表達方式:[A,B]mg/L、(A%,B%);線性下限準繩上不應為零,下限應契合臨床實踐需求。線性偏向可以分段給出相對偏向和相對偏向要求。設定相對偏向和相對偏向分界點時,準繩上應盡量將醫學決議水平濃度置于相對偏向要求區間內;且相對和相對偏向的要求應思索分界點的延續性。
舉例:醫學決議水平20mg/L,偏向要求中分界點為15mg/L,相對偏向要求≤1.5mg/L,相對偏向要求≤10%。
免疫類產品普通僅要求給出線性區間,可以不要求線性偏向。
162.產品技術要求中,檢出限/空白限如何要求?
答:免疫類產品普通有此項要求。可二選一,假設無相應國行標或指點準繩,建議給出檢出限要求。
163.產品技術要求中,準確度如何要求?
答:假設無相應國行標或指點準繩,準繩上采用三種方式之一:相對偏向(參考物質或參考方法定值物)、回收實驗(人源樣本中添加純品)、比對實驗(同類已上市產品)。關于有適用國度規范品的產品,應采用相對偏向方式。
164.產品技術要求中,重復性如何要求?
答:假設無相應國行標或指點準繩,可在線性區間范圍內,選擇2~3個不同濃度水平的樣本。濃度選擇時,應思索線性寬度以及醫學決議水平,可選正常值和異常值水平(建議為弱陽性)。
165.體外診斷試劑產品申報延續注冊時,應如何撰寫“產品概述”和“有關產品平安性、有效性主要評價內容”?
答:“產品概述”中建議論述:產品預期用途;主要組成成分;檢測原理;注冊證號及有效期。
“有關產品平安性、有效性主要評價內容”中建議論述:
(1)本次延續注冊進程中該產品無變化。
(2)若本次注冊證有效期內產品變卦狀況,應明白變卦次數、變卦時間及變卦事項。
(3)若本次醫療器械注冊證有效期內有新的醫療器械強迫性規范和/或國度規范品發布實施,為契合新的醫療器械強迫性規范和/或國度規范品所做變卦狀況。
(4)若本次延續注冊為注冊管理類別有調整的產品,應注明管理類別調整文件,并明白調整前管理類別和調整后管理類別。
(5)若本次延續注冊為分類編碼有調整的產品,應注明分類編碼調整文件,并明白調整前分類編碼和調整后分類編碼。
(6)若原醫療器械注冊證中載明要求繼續完成的事項,應說明事項的完成狀況。
(7)總結陳說本次延續注冊產品平安有效性,契合延續注冊要求。
166.目前,化學發光法、膠體金法、熒光法等封鎖系統的試劑添加與原注冊證說明書中適用儀器不同廠家的適用機型,能否依照《北京市醫療器械快速審評審批方法》第十三條申報快速審評審批?
答:思索到封鎖系統儀器適用性的效果,需求經過充沛驗證才干證明試劑在新增儀器上的平安有效性,建議注冊人添加適用儀器依照正常的“變卦注冊”流程及資料停止申報。
167.延續注冊體外診斷試劑產品為契合新的國度規范品發布實施所做的變化屬于無需操持變卦注冊手續或許無需變化的,應提供哪些申報資料?
答:(1)已注冊產品為契合新的國度規范品發布實施所做的變化屬于無需操持變卦注冊手續或許無需變化的說明;
(2)若有無需操持變卦注冊手續的變化,應提供相關證明資料(至少包括新舊國度規范品說明書復印件和新舊國度規范品說明書觸及條款對比表)。
168.契合《北京市醫療器械快速審評審批方法》第十三條的要求,申報快速審評審批的產品應預備哪些申報資料?
答:
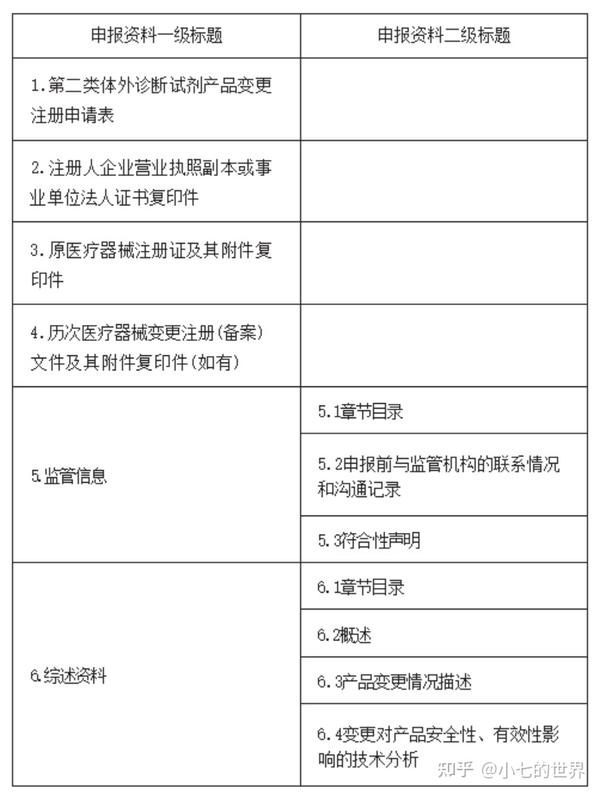

169.依照《北京市醫療器械快速審評審批方法》第十三條申報快速審評審批,添加適用儀器的,對所添加的適用儀器有什么要求?
答:(1)新增適用儀器應與原產品說明書中的適用儀器的自動化水平相反。
(2)新增適用儀器應與原產品說明書中的適用儀器為同類儀器。
(3)新增適用儀器應為國際已上市醫療器械,即已取得醫療器械注冊證。
以上三個條件同時滿足,才可以依照《北京市醫療器械快速審評審批方法》第十三條申報。
170.由III類降為II類的體外診斷試劑產品,在延續注冊進程中可以刪除《產品技術要求》中附錄里關于主要原資料、消費工藝及半成品要求的相關內容嗎?
答:依據《醫療器械產品技術要求編寫指點準繩》,關于第三類體外診斷試劑類產品,產品技術要求中應以附錄方式明白主要原資料、消費工藝要求。關于第二類體外診斷試劑類產品無此要求。
第四篇 醫療器械消費質量管理體系
171. 醫療器械注冊質量管理體系核對申報資料詳細包括哪些?要求是什么?
答:第二類醫療器械注冊質量管理體系核對的任務 流程及申報資料要求可登錄北京市藥品監視管理局網站( http://yjj.beijing.gov.cn/)查閱。
詳細文件包括:
(1)綜述注冊央求人應當承諾已依照相關法規要求樹立相應的質量管理體系,隨時接受質量管理體系核對。
(2)章節目錄應當包括本章的一切標題和小標題,注明目錄中各內容的頁碼。
(3)消費制造信息
A.產品描畫信息器械任務原理和總體消費工藝的簡明說明。
B.普通消費信息提供消費器械或其部件的一切地址和聯絡信息。如適用,應當提供外包消費、重要組件或原資料的消費(如植物組織和藥品)、關鍵工藝進程、滅菌等狀況的一切重要供應商稱號和地址。
(4)質量管理體系順序用于樹立和維護質量管理體系的高層級質量管理體系順序,包括質量手冊、質量方針、質量目的和文件及記載控制順序。
(5)管理職責順序用于經過論述質量方針、籌劃、職責/權限/溝通和管理評審,對樹立和維護質量管理體系構成管理保證文件的順序。
(6)資源管理順序用于為實施和維護質量管理體系所構成足夠資源(包括人力資源、基礎設備和任務環境)供應文件的順序。
(7)產品完成順序高層級的產品完成順序,如說明籌劃和客戶相關進程的順序。
A.設計和開發順序用于構成從項目初始至設計轉換的整個進程中關于產品設計的系統性和受控的開發進程文件的順序。
B.推銷順序用于構成契合已制定的質量和/或產品技術參數的推銷產品/效勞文件的順序。
C.消費和效勞控制順序用于構成受控條件下消費和效勞活動文件的順序,這些順序論述諸如產品的清潔和污染的控制、裝置和效勞活動、進程確認、標識和可追溯性等效果。
D.監視和測量裝置控制順序用于構成質量管理體系運轉進程中所運用的監視和測量設備已受控并繼續契合既定要求文件的順序。
(8)質量管理體系的測量、剖析和改良順序用于構成如何監視、測量、剖析和改良以確保產品和質量管理體系的契合性,并堅持質量管理體系有效性的文件的順序。
(9)其他質量體系順序信息不屬于上述內容,但對此次申報較為重要的其他信息。
(10)質量管理體系核對文件依據上述質量管理體系順序,注冊央求人應當構成相關質量管理體系文件和記載。應當提交下列資料,在質量管理體系核對時停止反省。
A.注冊央求人基本狀況表。
B.注冊央求人組織機構圖。
C.消費企業總平面布置圖、消費區域散布圖。
D.消費進程有污染要求的,應當提供有資質的檢測機構出具的環境檢測報告(附平面布局圖)復印件。
E.產品消費工藝流程圖,應當標明主要控制點與項目及主要原資料、推銷件的來源及質量控制方法。
F.主要消費設備和檢驗設備(包括進貨檢驗、進程檢驗、出廠*終檢驗所需的相關設備;在污染條件下消費的,還應當提供環境監測設備)目錄。
G.質量管理體系自查報告。
H.如適用,應當提供擬核對產品與既往已經過核對產品在消費條件、消費工藝等方面的對比說明。
第三類醫療器械注冊申報要求詳見國度藥品監視管理局網上辦事大廳(國度藥監局網上辦事大廳事項效勞搜索)http://zwfw.nmpa.gov.cn/web/tasklist/corporate?theme=YLQX。
172.醫療器械消費質量管理規范現場反省時間和反省員都有哪些要求?
答:(一)現場反省時間普通為1至3天;
(二)現場反省實行反省組長擔任制,反省組應當由2名以上(含2名)反省員組成。
(三)必要時,北京市藥品監視管理局可約請有關專家參與或委托第三方機構展開現場反省。
173.用于注冊的檢驗用產品和臨床實驗產品如何展開真實性核對?
答:用于注冊的檢驗用產品和臨床實驗產品的真實性核對與醫療器械注冊質量管理體系核對同時停止,重點查閱設計和開發進程實施籌劃和控制的相關記載、用于樣品消費的推銷記載、消費記載、檢驗記載和留樣觀察記載(如適用)等。
174.醫療器械產品變卦注冊能否會展開醫療器械注冊質量管理體系核對?
答:依據《醫療器械注冊管理方法》(國度市場監視管理總局令第47號)和《體外診斷試劑注冊管理方法》(國度市場監視管理總局令第48號)的規則,在對變卦注冊央求停止技術審評時,以為有必要對質量管理體系停止核對的,器械審查中心應當組織展開質量管理體系核對。
175.委托其他企業消費醫療器械的注冊人應當承當什么義務和責任?
答:1.依法承當醫療器械設計開發、臨床實驗、消費制造、銷售配送、售后效勞、產品召回、不良事情報告等環節中的相應法律責任。
2.與受托消費企業簽署委托合同和質量協議,明白委托消費中技術要求、質量保證、責任劃分、放行要求等責任,明白消費放行要求和產品上市放行方式。
3.增強對受托消費企業的監視管理,對受托消費企業的質量管理才干停止評價,活期對受托消費企業展開質量管理體系評價和審核。
4.增強不良事情監測,依據風險等級樹立醫療器械相應的追溯管理制度,確保醫療器械產品可滿足全程追溯的要求。
5.可以自行銷售醫療器械,也可以委托具有相關資質的醫療器械運營企業銷售。自行銷售的注冊人應當具有規則的醫療器械運營才干和條件;委托銷售的,應當簽署委托合同,明白各方權益義務。
6.經過信息化手腕,對研發、消費、銷售和不良事情監測狀況停止全流程追溯、監控。
7.確保提交的研討資料和臨床實驗數據真實牢靠、系統完整、可追溯。
176.委托受托消費企業消費醫療器械的注冊人應至少裝備哪些專業人員,需求滿足的要求是什么?
答:(1)應當裝備管理者代表。管理者代表受法定代表人或許主要擔任人委派,實行樹立、實施并堅持質量管理體系有效運轉等責任。
(2)應當裝備專門的研發技術人員,熟習所注冊醫療器械產品的研發和技術,具有相應的專業背景和任務閱歷,確保提交的研討資料和臨床實驗數據真實、完整、可追溯。
(3)應當裝備專門的質量管理人員,應具有任務閱歷,熟習所注冊醫療器械產品的消費質量管理要求,可以對醫療器械注冊人和受托消費企業的質量管理體系停止評價、審核和監視。
(4)應當裝備專門的法規事務人員,應具有任務閱歷,熟習所注冊醫療器械產品法規要求,可以處置相關法規事務。
(5)應當裝備專門的上市后事務人員,應具有任務閱歷,熟習醫療器械不良事情監測、產品召回、售后效勞等要求,可以處置相翻開市后事務。
177.國度藥品監視管理局《制止委托消費醫療器械目錄》觸及的產品能否可以經過注冊人委托受托消費企業展開消費活動?
答:對國度藥品監視管理局《制止委托消費醫療器械目錄》中的產品,注冊人不得經過委托受托消費企業的方式展開消費活動。
178.如何編制《醫療器械委托消費質量協議》?
答:委托消費是指醫療器械注冊人、備案人(以下稱“委托方”)委托其他消費企業(以下稱“受托方”)停止的消費活動。
經過《醫療器械委托消費質量協議》的簽署,明白醫療器械委托消費時,雙方在產品消費全進程中各自的權益、義務和責任;規范雙方對委托消費的醫療器械應當承當的產質量量平安義務和責任;保證委托消費的醫療器械契合注冊/備案和消費容許/備案的有關要求,實在保證上市醫療器械的平安、有效、質量可控,保證人體安康和生命平安。
詳細可參考國度藥品監視管理局發布的《醫療器械委托消費質量協議編制指南》。
179.注冊央求人能否可以委托其他機構展開醫療器械研發?
答:可以。注冊央求人應當明白產品研發活動委托的研發活動范圍、水平以及對受托研發機構的研發才干與繼續技術支持才干要求,并對受托方的研發才干與繼續技術支持才干停止評價,以確保其契合要求。
180.受托消費企業在消費受托消費的產品時,如何對自有產品與受托消費產品停止質量管理體系的控制?
答:受托消費企業有相反產品在產時,應當與受托消費產品有清楚區別的編號、批號及進程標識管理方式,防止混雜。
181.醫療器械注冊人應該如何展開委托消費,如何停止產品放行?
答:醫療器械注冊人、備案人委托消費的,應當對受托方的質量保證才干和風險管理才干停止評價,依照國度藥監局制定的委托消費質量協議指南要求,與其簽署質量協議以及委托協議,監視受托方實行有關協議商定的義務;受托消費企業應當依照法律、法規、規章、醫療器械消費質量管理規范、強迫性規范、產品技術要求、委托消費質量協議等要求組織消費,抵消費行為擔任,并接受醫療器械注冊人、備案人的監視。
受托消費企業應當向原消費容許或許消費備案部門報告添加消費的產品種類狀況,并提供委托方、受托消費產品、受托期限等信息;添加消費產品觸及消費條件變化,能夠影響產品平安、有效的,應當在添加消費產品30個任務日前向原消費容許部門報告,原消費容許部門應當及時展開現場核對。屬于容許事項變化的,應當依照規則操持相關容許變卦。
醫療器械注冊人、備案人應當擔任產品上市放行,樹立產品上市放行規程,明白放行規范、條件,并對醫療器械消費進程記載和質量檢驗結果停止審核,契合規范和條件的,經授權的放行人員簽字前方可上市。委托消費的,醫療器械注冊人、備案人還應當對受托消費企業的消費放行文件停止審核。受托消費企業應當樹立消費放行規程,明白消費放行的規范、條件,確認契合規范、條件的,方可出廠。不契合法律、法規、規章、強迫性規范以及經注冊或許備案的產品技術要求的,不得放行出廠和上市。醫療器械注冊人、備案人不得委托受托消費企業停止上市放行。
182.醫療器械的放行包括哪些方式?
答:醫療器械的放行包括消費放行和上市放行。
183.醫療器械注冊人、備案人、受托消費企業應該如何落實報告制度?
答:為進一步落實醫療器械注冊人、備案人、受托消費企業主體責任,《醫療器械消費監視管理方法》樹立醫療器械報告制度,并規則相應法律責任。醫療器械注冊人、備案人、受托消費企業應當依照規則執行醫療器械報告制度。
一是落實自查報告制度。醫療器械注冊人、備案人、受托消費企業應當每年對質量管理體系的運轉狀況停止自查,依照醫療器械消費質量管理體系年度自查報告編寫指南要求編寫自查報告,并于次年3月31日前向所在地藥品監視管理部門提交自查報告。出口醫療器械注冊人、備案人由其代理人向代理人所在地省、自治區、直轄市藥品監視管理部門提交自查報告。
二是落實消費產品種類報告制度。醫療器械消費企業應當向藥品監視管理部門報告所消費的產品種類狀況。添加消費產品種類的,應當向原消費容許或許消費備案部門報告,觸及委托消費的,還應當提供委托方、受托消費產品、受托期限等信息。醫療器械消費企業添加消費產品觸及消費條件變化,能夠影響產品平安、有效的,應當在添加消費產品30個任務日前向原消費容許部門報告,原消費容許部門應當及時展開現場核對。屬于容許事項變化的,應當依照規則操持相關容許變卦。
三是落實消費條件變化報告制度。醫療器械注冊人、備案人、受托消費企業的消費條件發作變化,不再契合醫療器械質量管理體系要求的,應當立刻采取整改措施;能夠影響醫療器械平安、有效的,應當立刻中止消費活動,并向原消費容許或許消費備案部門報告。受托消費企業應當及時將變化狀況告知醫療器械注冊人、備案人。
四是落實重重消費報告制度。醫療器械消費企業延續停產一年以上且無同類產品在產的,重重消費時,應當停止必要的驗證和確認,并書面報告藥品監視管理部門。能夠影響質量平安的,藥品監視管理部門可以依據需求組織核對。
184.醫療器械注冊央求人樹立質量管理體系文件的詳細要求都有哪些?
答:央求人應當樹立與申報注冊產品相順應的質量管理體系文件,包括質量手冊、順序文件、技術文件和數據記載等。技術文件應當包括產品技術要求及相關規范、消費工藝規程、作業指點書、檢驗和實驗操作規程等相關文件。數據、記載應當確保產品設計開發、物料推銷、消費、質量控制、產品放行等活動的可追溯。
185.生物平安柜在運用中需求留意什么??
答:生物平安柜裝置完成,位置移動后需停止裝置檢驗。每年至少停止一次維護檢驗,當生物平安柜改換過濾器和外部部件維修后,也要停止維護檢驗。詳細檢驗要求和檢驗項目見《生物平安柜》(YY0569)。
186.無菌和植入性醫療器械消費企業能否可以對產品停止參數放行?
答:關于自行具有產品滅菌才干的無菌、植入性醫療器械消費企業,其出廠檢驗項目規則對產品停止無菌檢驗,可依照規則執行;如規則檢測生物指示劑(菌片),在對滅菌進程停止迷信驗證基礎上,可采取檢測菌片方式停止產品放行。
187.非無菌形狀提供的醫療器械需求在潔凈環境下組織消費嗎?
答:非無菌形狀提供的植入性醫療器械,其末道清洗和包裝工序的消費環境應當參照《無菌醫療用具消費管理規范》(YY0033-2000)規范或自行驗證并確定消費環境,有效控制初始污染菌。
188.醫療器械注冊央求人全項目委托有資質的醫療器械檢驗機構,能否適用《北京市醫療器械注冊自檢質量管理體系現場反省指點準繩(試行)》的要求?
答:不適用。
189.醫療器械注冊核對進程中,能否要求注冊央求人布置靜態消費?
答:在現場反省時,注冊央求人相關人員應當熟習并可以完成產品的相關操作,相關設備應當可以正常運轉,以證明注冊央求人具有申報產品的消費才干。關于申報無菌、植入性醫療器械產品注冊時展開的注冊核對,應當展開靜態消費。
190.醫療器械注冊質量管理體系核對現場反省的結果都有哪些?如需整改的,整改期限是多久?
答:醫療器械注冊質量管理體系核對中,反省組對現場反省提出的建議結論分為“經過反省”、“整改后復查”、“未經過反省”三種狀況。建議結論為“經過反省”或“未經過反省”的,市器械審查中心自現場反省完畢后5個任務日內對反省組提交的反省資料停止審核,提出審查結論。建議結論為“整改后復查”的,注冊央求人在現場反省完畢后1個月內,一次性向市器械審查中心提交整改報告和復查央求,市器械審查中心自收到反省組報送建議結論和反省資料,以及注冊央求人提交的整改報告和復查央求后,5個任務日內停止審核,提出審查結論。審查結論仍為“整改后復查”的,市器械審查中心應當自作出審查結論之日起3個任務日內將需求整改的內容告知注冊央求人。注冊央求人自收到整改意見之日起 5 個月內一次性向市器械審查中心提交整改報告和復查央求。市器械審查中心在收到整改報告和復查央求后30個任務日內完成復查。可以經過資料停止核實的,可免于現場復查。整改后經過核對的,審查結論為“整改后經過核對”。未在規則期限內提交復查央求和整改報告的,或整改復查后仍達不到“經過核對”要求的,審查結論為“整改后未經過核對”。注冊央求人拒絕接受體系核對的,審查結論為“未經過核對”。
第五篇 分類界定
191. 目前,可以參考的體外診斷試劑分類界定文件有 哪些?
答:截止2022年7月31日,可以參考的體外診斷試劑分類界定文件如下:
(1)《體外診斷試劑分類規則》
(2)《6840體外診斷試劑分類子目錄(2013版)》
(3)總局關于過敏原類、流式細胞儀配套用、免疫組化和原位雜交類體外診斷試劑產品屬性及類別調整的通告(2017年第226號)的附件《流式細胞儀配套用體外診斷試劑產品分類列表》《免疫組化和原位雜交類體外診斷試劑產品分類列表》《不作為醫療器械管理產品列表》
(4)國度藥監局關于調整《6840體外診斷試劑分類子目錄(2013版)》局部外容的公告(2020年第112號)
(5)2018年至今共7次醫療器械產品分類界定結果匯總
192.關于新研制未列入《醫療器械分類目錄》的醫療器械能否一定需求停止分類界定后才干操持注冊或備案?
答:否。依據《醫療器械監視管理條例》第二十三條規則對新研制的尚未列入分類目錄的醫療器械,央求人可以依照本條例有關第三類醫療器械產品注冊的規則直接央求產品注冊,也可以依據分類規則判別產品類別并向國務院藥品監視管理部門央求類別確認后依照本條例的規則央求產品注冊或許停止產品備案。
193.申報醫療器械分類界定前,央求人需求完成哪些任務?
答:一是產品設計開發任務已基本完成,產品已定型,有樣機或產品,擬展開央求產品注冊或備案的相關任務。
二是停止屬性界定,依據醫療器械定義,判定產品能否屬于醫療器械,若經判定屬于醫療器械停止后續事宜。
三是查閱相關分類界定文件,經過對照產品稱號、預期用途、結構組成、運用方法等多方面綜合判定,確認擬央求分類界定產品確不在已地下的一切分類界定文件中。
四是查找國際外能否有已上市的同類或近似產品。
五是依照《北京市醫療器械分類界定申報要求》預備相關技術文檔。
194.央求醫療器械分類界定,央求人需求完成哪些申報任務?
答:*步:央求人經過中國食品藥品檢定研討院官網首頁→辦事大廳→醫療器械規范與分類管理→醫療器械分類界定信息系統(網址:http://app.nifdc.org.cn/biaogzx)停止網絡注冊,注冊后填寫《分類界定央求表》,并上傳其他央求資料。
第二步:央求人應將在線打印的《分類界定央求表》,連同其他央求資料加蓋央求人騎縫章,將央求資料經過郵寄或現場方式提交至北京市醫療器械審評反省中心。(地址:北京市西城區水車胡同13號,聯絡電話:010-58549949)。
紙質版央求資料應契合《北京市醫療器械分類界定申報要求》,同時保證紙質央求資料與網上填報資料的分歧性。
195.網上填報時有哪些留意事項?
答:(1)《醫療器械產品分類界定注銷表》內容填寫完整準確,不適用的項目應注明“無”或“不適用”,請勿空項。
(2)《醫療器械產品分類界定注銷表》中“作用原理或機理”項應填寫清楚,例如液體敷料產品,應列表寫明每個成分及成分的作用,否則無法判定其屬性或類別。
(3)《醫療器械產品分類界定注銷表》中“國際外近似產品”項如有請填寫,應寫明擬申報產品與國際外近似產品的異同點,并說明近似產品的分類狀況。
(4)《醫療器械產品分類界定注銷表》中“企業意見”項必需填寫,明白企業主張和界定類別,同時撰寫企業主張理由,理由可參考分類界定相關文件,也可參考已上市同類產品狀況。
(5)網上上傳的附件應完整,主張創新的產品應上傳相關屬于新產品的資料。
196.預備紙質版資料時有哪些留意事項?
答:(1)《醫療器械產品分類界定注銷表》企業的法定代表人應簽字,填寫申報日期并加蓋公章。
(2)有營業執照的企業應提供營業執照正本復印件以證明公司法定代表人狀況。
(3)紙質版資料都應加蓋公章,非單頁資料還應加蓋騎縫章。
(4)《醫療器械產品分類界定注銷表》中聯絡人若不是法定代表人應當提交《授權委托書》。
197.境內產品與出口及港、澳、臺產品在央求醫療器械分類界定時詳細操作有何不同?
答:境內產品的相關資料提交給至央求企業所在地的省級藥品監視管理部門,出口及港、澳、臺產品的相關資料提交至國度藥品監視管理局醫療器械規范管理中心。
198.央求分類界定時提交的產品技術要求和產品說明書應契合哪些要求?
答:產品技術要求應契合《醫療器械產品技術要求編寫指點準繩》;醫療器械產品說明書應契合《醫療器械說明書和標簽管理規則》,體外診斷試劑說明書應契合《醫療器械說明書和標簽管理規則》以及《體外診斷試劑說明書編寫指點準繩》。
199.紙質版資料中需求提交《授權委托書》和《自我保證聲明》,請問相關資料有模板嗎?
答:有模板,詳細下載地址為:http://yjj.beijing.gov.cn/yjj/zwhd/bgxz/ylqx/index.html。
200.目前,可以參考的醫療器械分類界定文件有哪些?
答:截止2022年7月31日,可以參考的醫療器械分類界定文件如下:












