無處不在的細菌如何被檢測 ?無處不在的細菌如何被檢測 ?健明迪
無處不在的細菌如何被檢測??無處不在的細菌如何被檢測??健明迪
陰道微生態檢測,究竟檢測什么呢?

陰道微生態檢測是陰道炎癥診斷的搶手新詞,如今很多大醫院也都有了陰道微生態檢測這個項目。那么陰道微生態檢測究竟檢測什么呢?


普通來說陰道微生態檢測分為形狀學檢測和功用學檢測。
形狀學檢測:包括菌群密集度、菌群多樣性、優勢菌、滴蟲、真菌等。
功用學檢測:包括PH值、過氧化氫、唾液酸苷酶、白細胞酯酶、?-葡萄糖醛酸苷酶、乙酰氨基糖苷酶等。
陰道微生態診斷方案為形狀學結合功用學檢測,形狀學檢測為主,功用學檢測為輔。若形狀學檢測與功用學檢測結果不分歧時,目前以形狀學檢測為主要參考目的。
VVC和TV診斷相對比擬復雜,普統統過對白帶的顯微鏡觀察,找到病原可以診斷,但AV、BV、CV、DV則復雜得多,單純從形狀上很難找到病原,更多的是從白帶的一些功用反省來確診。
1、形 態 學 檢 測
檢測方法:鏡檢法,油鏡下反省陰道菌群。
菌群密集度:
依據100倍油鏡下每個視野的平均細菌數分為Ⅰ~Ⅳ級。Ⅰ級:1~10個菌/油鏡;Ⅱ級:10~100個菌/油鏡;Ⅲ級:100~1000個菌/油鏡;Ⅳ級:1000以上/油鏡。
正常為Ⅱ~Ⅲ級,Ⅳ級為菌群增殖過度。
菌群多樣性:
依據100倍油鏡下視野區分出的細菌菌群數目可分為Ⅰ~Ⅳ級。Ⅰ級:1~3種菌群/油鏡;Ⅱ級:4~6種菌群/油鏡視野;Ⅲ級:7~9種菌群/油鏡;Ⅳ級:10種以上菌群/油鏡。
正常為Ⅱ~Ⅲ級。
優勢菌:
油鏡下所見*多的微生物可以定義為優勢菌, 正常應檢出革蘭陽性桿菌。
菌群抑制:缺少優勢菌, 陰道菌群多樣性≤Ⅰ級。
菌群增殖過度:優勢菌為乳酸桿菌, 但菌群密集度出現增高, 為Ⅳ級。
病原微生物:
主要是經過觀察真菌菌絲和 (或) 滴蟲,顯微鏡鏡檢陰道分泌物中能否存在滴蟲或真菌假菌絲、芽生孢子、孢子等。
①真菌檢測:油鏡下可發現真菌卵圓形孢子、芽生孢子或管狀的假菌絲,革蘭染色陽性。當鏡檢發現芽生孢子或假菌絲時,應報告為外陰陰道假絲酵母菌病(VVC)。
②滴蟲檢測:革蘭染色陽性,較白細胞略大,形狀不規則,內有食物泡,周邊有少量的白細胞或上皮細胞碎片,發現滴蟲,可診斷為滴蟲陰道炎。
2、功 能 學 檢 測


檢測方法:檢測需氧菌、厭氧菌、真菌、滴蟲等的代謝產物、酶的活性及pH值。
pH值:
正常狀況下由于乳酸桿菌產酸,正常陰道內的pH≤4.5,多在3.8~4.4,細菌性陰道病患者的陰道pH值普通大于4.5。
過氧化氫:
陽性代表乳酸桿菌發生的過氧化氫濃度降低,陰道內乳酸桿菌增加,陰性代表乳酸桿菌正常。
唾液酸苷酶:
這是惹起細菌性陰道病的主要厭氧菌釋放的,唾液酸酐酶呈陽性,意味著厭氧菌感染。是細菌性陰道病的表現。
白細胞酯酶:
以前僅僅依據白細胞數量判別炎癥,目前要看白細胞吞噬細菌的功用。陰道內有炎癥時,白細胞釋放白細胞酯酶,白細胞酯酶增高,化驗單上就標注為陽性。白細胞酯酶陽性才代表陰道炎,而白細胞增多,白帶清潔度Ⅲ~Ⅳ度并不能診斷炎癥。
凝結酶、?-葡萄糖醛酸苷酶:
這是需氧菌釋放的,這兩個目的之一陽性,意味著需氧菌性陰道炎。
乙酰氨基糖苷酶:
是白色念珠菌的特異性酶,一旦其水平出現降低,提示患者存在陰道內膜損傷。
陰道微生態是經過這些復雜的目的反省,*后經過軟件剖析停止診斷。
3、分 子 檢 測
慣例鏡檢法是臨床診斷陰道炎病原體的常用手腕,但診斷結果會遭到諸多要素影響,如分泌物涂片質量、氣溫、判別規范和檢測人員閱歷不分歧等,影響診斷準確性,容易漏檢,誤判。
而微生物的代謝酶數量眾多, 酶學檢測的效果在于, 酶的陽性與否并不是陰道炎診斷的必要要素, 以BV為例, 大約90%的BV患者會出現唾液酸酶陽性, 但是唾液酸酶陽性的患者并不一定都是BV。因此, 酶學檢驗只能作為形狀學檢驗的重要補充, 有助于我們判別感染的嚴重水平以及混合感染等特殊狀況。
牢靠的檢測技術在婦科感染疾病診治中占有十分重要的位置,分子生物學技術的出現、精準醫學時代的到來,為臨床上這些診治困難的感染性疾病帶來了新曙光、在微生物研討范圍中具有里程碑式的意義。
經過對30種相關生殖道有益/致病微生物的精準基因檢測,一次性更精準、片面的了解陰道微生態,對陰道菌群停止生態分型評價。其高特異性、高靈敏度是替代傳統鏡檢/培育法的有效提高。微生物鑒定與定量檢驗并行,提醒生殖道感染治病機理,協助女性更精準的了解陰道菌群結構。
無處不在的細菌如何被檢測?
作為微生物中一個重要的研討范圍,細菌,與我們的安康和生活親密相關,其菌種類單一,體積龐大,結構不同。在環境范圍、食品范圍、臨床醫學等,經常需求對細菌種類、細菌數量、細菌中活性物質、細菌作為媒介分解化合物、單個活性菌體等停止快速檢測和剖析。
與慣例檢測技術相比,顯微拉曼光譜是一種快速、無損、無懼水、微區分辨、非接觸式檢測的分子光譜技術,十分適宜用于微生物的檢測和剖析,成像功用還可以完成細菌微區成分散布的剖析。
儀器和軟件
Thermo Scientific ? DXR? 3系列顯微拉曼技術在微生物檢測和剖析范圍具有共同的技術和運用優勢:
先進的自動化和智能化光學控制系統
超高的檢測靈敏度和空間分辨率
軟件控制、實時到樣品的激光功率精細調理專利技術
“一鍵式”操作,無需專業背景
功用弱小、界面友好的剖析軟件
集成數學模型算法、導游操作的TQ Analyst建模軟件
光鑷-微流控聯用技術完成流體中微生物的快速剖析
運用案例
細菌結構剖析
細菌種類不同,其活性物質如氨基酸、蛋白質等表現出結構和量的差異,拉曼光譜的特征結構峰可以很好的表現活性物質和含量的差異,依據這些差異可以快速無損的停止細菌種類的鑒別和剖析。
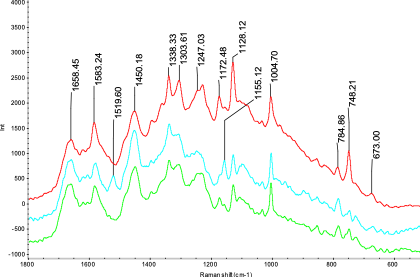
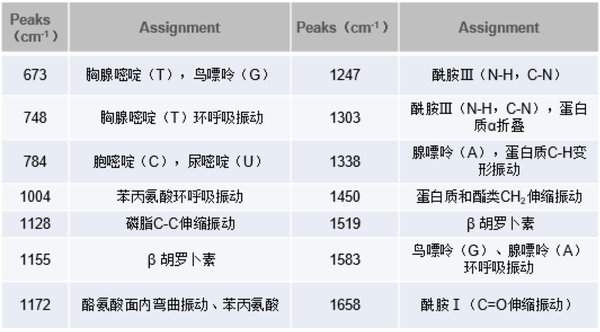
單個細菌檢測
通常細菌的體積較小,信號較弱,溶液中細菌濃度較低時,很難檢測到有效的化學結構信息,尤其是需求對單個細菌的化學結構停止檢測和剖析。在停止分子結構剖析時,需求高靈敏度和空中間分辨率的拉曼技術。
采用超高靈敏和空中間分辨的DXRxi顯微拉曼成像光譜儀,應用研討者[1]分解的磁性富集高活性SERS傳感器,經過適配子識別細菌,完成低濃度和單個金黃色葡萄球菌的檢測和剖析。
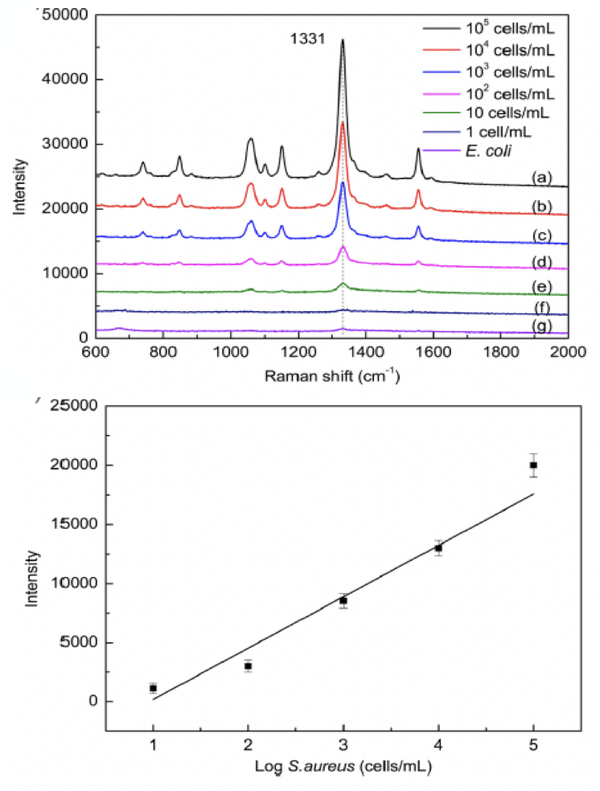
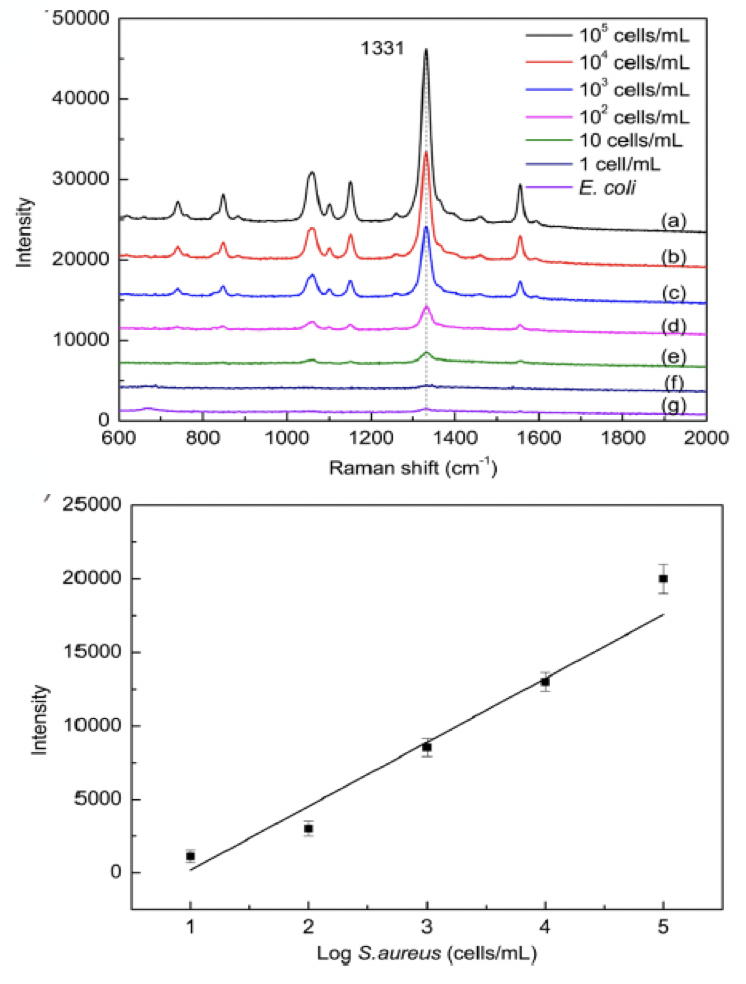
單個細菌體內分解物的鑒別與剖析
酵母菌是一些單細胞真菌,其結構與初等植物的細胞一樣,比擬復雜,體積小的約2-6um,大的約5-30um。在一定適宜的培育條件下,酵母菌菌體可以作為微型“反響器”,在體內分解一些目的化合物,在剖析目的化合物時,需求空中間分辨率的顯微拉曼光譜儀才干實如今菌體微區的剖析,同時要求快速測試以堅持細菌的活性。而賽默飛的DXR3xi拉曼成像光譜儀因其超高靈敏度、空中間分辨率和超快速成像功用,是一款十分適宜用于這些范圍的研討。
Thermo Scientific ?DXR?系列顯微拉曼光譜儀,經過蛋白質、氨基酸等生物活性物質的拉曼光譜特征性,可以輕松完成幾微米甚至幾百納米大小細菌種類的檢測與剖析。應用超高靈敏度和超空中間分辨的DXR3xi成像技術有助于快速研討單個活性細菌體內物質的散布,完成微生物的活體剖析。為微生物的研討提供了一種無損、快速、無懼水的研討工具。
參考文獻
1,Junfeng Wang et al, ACS Appl. Mater. Interfaces 2015, 7, 20919?20929
無處不在的細菌如何被檢測??無處不在的細菌如何被檢測??健明迪
SignalP+TMHMM預測微生物分泌蛋白
Secretory Protein是指在細胞內分解后,分泌到細胞外起作用的蛋白質。分泌蛋白的N 端有普通由15~30 個氨基酸組成的信號肽。信號肽是引導新分解的蛋白質向分泌通路轉移的短(長度5-30個氨基酸)肽鏈。常指新分解多肽鏈中用于指點蛋白質的跨膜轉移(定位)的N-末端的氨基酸序列(有時不一定在N端)。運用SignalP 注釋蛋白序列能否含有信號肽結構,運用TMHMM注釋蛋白序列能否含有跨膜結構,*終挑選出含有信號肽結構并且不含跨膜結構的蛋白為分泌蛋白。
軟件Software
- SignalP V6.0
- SignalP 6.0 預測來自古細菌、革蘭氏陽性細菌、革蘭氏陰性細菌和真核生物的蛋白質中存在的信號肽predicts signal peptides and the location of their cleavage sites in proteins from Archaea, Gram-positive Bacteria,及其切割位點的位置。Gram-negative Bacteria and Eukarya.在細菌和古細菌中,SignalP 6.0 可以區分五種類型的信號肽:In Bacteria and Archaea, SignalP 6.0 can discriminate between five types of signal peptides:
- Sec/SPI:由 Sec 轉座轉運,并由信號肽酶 I (Lep) 切割的“規范”分泌信號肽;"Standard" secretory signal peptides transported by Sec translocon and cleaved by Signal Peptidase I (Lep).
- Sec/SPII:由 Sec 轉座子運輸,并由信號肽酶 II (Lsp) 切割的脂蛋白信號肽;lipoprotein signal peptides transported by the Sec translocon and cleaved by Signal Peptidase II (Lsp).
- Tat/SPI:由 Tat 轉座子轉運,并由信號肽酶 I (Lep) 切割的 Tat 信號肽;Tat signal peptides transported by the Tat translocon and cleaved by Signal Peptidase I (Lep).
- Tat/SPII:由 Tat 轉位子轉運,并由信號肽酶 II (Lsp) 切割的 Tat 脂蛋白信號肽;Tat lipoprotein signal peptides transported by Tat translocon & cleaved by Signal Peptidase II (Lsp).
- Sec/SPIII:由 Sec 轉位子運輸,并由信號肽酶 III (PilD/PibD) 切割的菌毛蛋白和菌毛蛋白樣信號肽。Pilin & pilin-like signal peptides transported by Sec translocon & cleaved by Signal Peptidase III (PilD/PibD).
- 此外,SignalP 6.0 預測信號肽的區域。Additionally, SignalP 6.0 predicts the regions of signal peptides.依據類型,預測 n、h 和 c 區域以及其他顯著特征的位置。Depending on the type, the positions of n-, h- and c-regions as well as of other distinctive features are predicted.
- TMHMM V2.0c
- 用于預測蛋白質中的跨膜螺旋。
- Python
SignalP和TMHMM關于學術用戶收費,但是需求填寫相關信息和郵箱,以接納下載鏈接(4h有效時間)。
軟件裝置Installation of Softwares
裝置SignalP 6.0
- 下載 訪問SignalP V6.0網站,找到“Download”,填寫相關信息,獲取下載鏈接,下載失掉“signalp-6.0.fast.tar.gz”。有兩個形式可以選擇——“slow_sequential”和“fast"。前者runs the full model sequentially, taking the same amount of RAM as
fastbut being 6 times slower;后者uses a smaller model that approximates the performance of the full model, requiring a fraction of the resources and being significantly faste。本教程下載的是fast形式。 - 裝置Installation
- 裝置依賴Dependencies
- Python
- matplotlib>3.3.2
- numpy>1.19.2
- torch>1.7.0 pip install torch
- tqdm>4.46.1
- 裝置SignalP 6.0 # 解緊縮裝置文件 tar zxvf signalp-6.0.fast.tar.gz # 進入解壓后的軟件目錄,在終端運轉 python setup.py install # 測試裝置 signalp6 --help
裝置TMHMM V2.0c
- 下載 訪問TMHMM V2.0c網站,找到“Download”,填寫相關信息,獲取下載鏈接,下載失掉“tmhmm-2.0c.Linux.tar.gz”。
- 裝置 # 解緊縮 tar zxvf tmhmm-2.0c.Linux.tar.gz # 進入解壓后的目錄 cd tmhmm-2.0c # 獲取以后途徑,我的是“/home/liu/tools/tmhmm-2.0c/bin” pwd # 將該途徑參與到系統的環境變量中,參考我之前的文章來(編輯~/.bashrc)http://liaochenlanruo.github.io/post/f6c9.html#%E6%B7%BB%E5%8A%A0%E7%8E%AF%E5%A2%83%E5%8F%98%E9%87%8F # 修正bin目錄下的tmhmm和tmhmmformat.pl的首行為“#!/usr/bin/perl”
- 運轉錯誤 運轉軟件時總報
Segmentation fault (core dumped)錯誤,暫時無解。各位可以運用其在線版。
軟件用法Usage
SignalP 6.0
預測Prediction
A command takes the following form
signalp6 --fastafile /path/to/input.fasta --organism other --output_dir path/to/be/saved --format txt --mode fast
fastafile輸入文件為FASTA格式的蛋白序列文件Specifies the fasta file with the sequences to be predicted.。organismis eitherotherorEukarya. SpecifyingEukaryatriggers post-processing of the SP predictions to prevent spurious results (only predicts type Sec/SPI).formatcan take the valuestxt,png,eps,all. It defines what output files are created for individual sequences.txtproduces a tabular.gfffile with the per-position predictions for each sequence.png,eps,alladditionally produce probability plots in the requested format. For larger prediction jobs, plotting will slow down the processing speed significantly.modeis eitherfast,sloworslow-sequential. Default isfast, which uses a smaller model that approximates the performance of the full model, requiring a fraction of the resources and being significantly faster.slowruns the full model in parallel, which requires more than 14GB of RAM to be available.slow-sequentialruns the full model sequentially, taking the same amount of RAM asfastbut being 6 times slower. If the specified model is not installed, SignalP will abort with an error.
輸入Outputs
- output_dir/output.gff3:僅包括含有信號肽的序列信息;
- output_dir/prediction_results.txt:包括了輸入文件中的一切序列(不重要);
- output_dir/region_output.gff3:包括一切的信號肽區域信息。
- n-region: The n-terminal region of the signal peptide. Reported for Sec/SPI, Sec/SPII, Tat/SPI and Tat/SPII. Labeled as N
- h-region: The center hydrophobic region of the signal peptide. Reported for Sec/SPI, Sec/SPII, Tat/SPI and Tat/SPII. Labeled as H
- c-region: The c-terminal region of the signal peptide, reported for Sec/SPI and Tat/SPI.
- Cysteine: The conserved cysteine in +1 of the cleavage site of Lipoproteins that is used for Lipidation. Labeled as c.
- Twin-arginine motif: The twin-arginine motif at the end of the n-region that is characteristic for Tat signal peptides. Labeled as R.
- Sec/SPIII: These signal peptides have no known region structure.
批處置與結果優化
腳本名:run_SignalP.pl
#!/usr/bin/perl
use strict;
use warnings;
# Author: Liu Hualin
# Date: Oct 14, 2021
open IDNOSEQ, ">IDNOSEQ.txt" || die;
my @faa = glob("*.faa");
foreach (@faa) {
$_ =~ /(.+).faa/;
my $str = $1;
my $out = $1 . ".nodesc";
my $sigseq = $1 . ".sigseq";
my $outdir = $1 . "_signalp";
open IN, $_ || die;
open OUT, ">$out" || die;
while (
chomp;
if (/^(>\S+)/) {
print OUT $1 . "\n";
}else {
print OUT $_ . "\n";
}
}
close IN;
close OUT;
my %hash = idseq($out);
system("signalp6 --fastafile $out --organism other --output_dir $outdir --format txt --mode fast");
my $gff = $outdir . "/output.gff3";
if (! -z $gff) {
open IN, "$gff" || die;
open OUT, ">$sigseq" || die;
while (
chomp;
my @lines = split /\t/;
if (exists $hash{$lines[0]}) {
print OUT ">$lines[0]\n$hash{$lines[0]}\n";
}else {
print IDNOSEQ $str . "\t" . "$lines[0]\n";
}
}
close IN;
close OUT;
}
system("rm $out");
system("mv $sigseq $outdir");
}
close IDNOSEQ;
sub idseq {
my ($fasta) = @_;
my %hash;
local $/ = ">";
open IN, $fasta || die;
while (
chomp;
my ($header, $seq) = split (/\n/, $_, 2);
$header =~ /(\S+)/;
my $id = $1;
$hash{$id} = $seq;
}
close IN;
return (%hash);
}
將run_SignalP.pl與后綴名為“.faa”的FASTA格式文件放在同一目錄下,在終端中運轉如下代碼:
perl run_SignalP.pl
結果解讀Output interpretation
*代表輸入文件的名字。
- *_signalp/output.gff3:僅包括含有信號肽的序列信息;
- *_signalp/prediction_results.txt:包括了輸入文件中的一切序列(不重要);
- *_signalp/region_output.gff3:包括一切的信號肽區域信息;
- *_signalp/*.sigseq:存儲一切信號肽的氨基酸序列文件,可用作TMHMM的輸入文件。
TMHMM
預測
離線版總是報錯,找不出緣由,因此運用網頁效勞器停止,輸入文件為上述生成的“*_signalp/*.sigseq”,將其上傳至網頁版TMHMM,提交義務,等候結果即可。
結果展現
TMHMM可以輸入多種格式的結果文件,詳細請參考其官方說明。
在TMHMM網站提交義務
- Long output format
- Length: 蛋白序列的長度。The length of the protein sequence.
- Number of predicted TMHs:預測到的跨膜螺旋的數量。The number of predicted transmembrane helices.
- Exp number of AAs in TMHs:跨膜螺旋中氨基酸的預期數量。The expected number of amino acids intransmembrane helices. 假設此數字大于 18,則很能夠是跨膜蛋白(或具有信號肽)。If this number is larger than 18 it is very likely to be a transmembrane protein (OR have a signal peptide).
- Exp number, first 60 AAs:在蛋白的前60個氨基酸中跨膜螺旋中氨基酸的預期數量。The expected number of amino acids in transmembrane helices in the first 60 amino acids of the protein.假設該數字超越幾個,你應該被正告在 N 端預測的跨膜螺旋能夠是一個信號肽。If it more than a few, you are warned that a predicted transmembrane helix in the N-term could be a signal peptide.
- Total prob of N-in:N端在膜的細胞質一側的總概率。The total probability that the N-term is on the cytoplasmic side of the membrane.
- POSSIBLE N-term signal sequence:當“Exp number, first 60 AAs”大于 10 時發生的正告。A warning that is produced when "Exp number, first 60 AAs" is larger than 10.
- 蛋白F01_bin.1_00110合計436個氨基酸,有5個跨膜螺旋結構。
- 蛋白F01_bin.1_00142合計557個氨基酸,一切序列均在膜外,即該序列編碼的是分泌蛋白。
- Short output format
- "len=": 蛋白序列的長度。The length of the protein sequence.
- "ExpAA=":跨膜螺旋中氨基酸的預期數量。The expected number of amino acids intransmembrane helices.假設此數字大于 18,則很能夠是跨膜蛋白(或具有信號肽)。If this number is larger than 18 it is very likely to be a transmembrane protein (OR have a signal peptide).
- "First60=":在蛋白的前60個氨基酸中跨膜螺旋中氨基酸的預期數量。The expected number of amino acids in transmembrane helices in the first 60 amino acids of the protein.假設該數字超越幾個,你應該被正告在 N 端預測的跨膜螺旋能夠是一個信號肽。If it more than a few, you are warned that a predicted transmembrane helix in the N-term could be a signal peptide.
- "PredHel=":預測到的跨膜螺旋的數量。The number of predicted transmembrane helices by N-best.
- "Topology=":N-best 預測的拓撲結構。The topology predicted by N-best.拓撲是由跨膜螺旋的位置給出的,假設螺旋在外部,則由“i”分隔,假設螺旋在外部,則由“o”分隔。'i7-29o44-66i87-109o'意味著它從膜內末尾,在位置7到29有一個預測的TMH,30-43在膜外,然后是位置44-66的TMH。
結果匯總
經過網頁版預測我們僅失掉了一個列表文件(Short output format),該文件需求自己復制網頁內容粘貼到新文件中,我將其命名為*_TMHMM_SHORT.txt,并將其寄存在*_signalp目錄中,該目錄是由run_SignalP.pl生成的。下面我將會統計各個基因組中信號肽蛋白的總數量、分泌蛋白數量和跨膜蛋白數量到文件Statistics.txt中,并區分提取每個基因組的分泌蛋白序列到*_signalp/*.secretory.faa文件中,提取跨膜蛋白序列到*_signalp/*.membrane.faa文件中。該進程將經過tmhmm_parser.pl完成。
#!/usr/bin/perl use strict; use warnings; # Author: Liu Hualin # Date: Oct 15, 2021 open OUT, ">Statistics.txt" || die; print OUT "Strain name\tSignal peptide numbers\tSecretory protein numbers\tMembrane protein numbers\n"; my @sig = glob("*_signalp"); foreach my $sig (@sig) { $sig=~/(.+)_signalp/; my $str = $1; my $tmhmm = $sig . "/$str" . "_TMHMM_SHORT.txt"; my $fasta = $sig . "/$str" . ".sigseq"; my $secretory = $str . ".secretory.faa"; my $membrane = $str . ".membrane.faa"; open SEC, ">$secretory" || die; open MEM, ">$membrane" || die; my $out = 0; my $on = 0; my %hash = idseq($fasta); open IN, $tmhmm || die; while (
運轉方法:將tmhmm_parser.pl放在*_signalp的上一級目錄下,*_signalp目錄中必需包括*_TMHMM_SHORT.txt文件和*.sigseq文件。在終端運轉如下代碼:
perl tmhmm_parser.pl
腳本獲取
本文腳本見GitHub。
敬告:運用文中腳本請援用本文網址,請尊重自己的休息效果,謝謝!Notice: When you use the scripts in this article, please cite the link of this webpage. Thank you!
參考
原文鏈接:SignalP+TMHMM預測微生物分泌蛋白 | liaochenlanruo
轉載請注明出處!











